

Genome Research-闫致强团队解析KCNQ4遗传性耳聋基因4085个突变的功能,为其基因诊断等提供依据
2022年6月27日,Genome Research杂志在线发表了闫致强课题组的研究论文“Proactive functional classification of all possible missense single-nucleotide variants in KCNQ4”。该研究前瞻性的通过膜片钳技术检测了遗传性耳聋基因KCNQ4所有可能的4,085个错义SNVs的电生理功能,提供功能证据支持解析已发现的意义不明和新发现的KCNQ4错义突变的致病性,对KCNQ4致病突变体携带者的生育判断和健康维护至关重要,体现了很强的临床应用价值。
全外显子组测序已经产生了海量的数据,检测出大量的疾病相关的变异,但对这些测序数据的发掘和变异的解释还是远远不够的,远不及变异发现的速度,是目前临床上急需解决的问题之一[1]。临床测序结果中发现大多数的变异属于错义单核苷酸突变(single-nucleotide variants, SNVs)且意义不明(variants of uncertain significance, VUS),给临床诊断和治疗带来很大的困扰。本研究以遗传性耳聋基因KCNQ4为研究切入点,进行大规模的错义突变的功能测试。已有研究表明,人类电压门控钾通道基因KCNQ4的功能缺失突变会引起常染色体显性非综合征型听力损失,一种典型的迟发性的语后渐进性耳聋(DFNA2)[2]。KCNQ4是迄今发现的最常见的引起非综合征型听力损失的基因之一,根据gnomAD数据库显示,大约每100人中有1人携带KCNQ4错义突变,且大多数致病性不明。在已经鉴定的DFNA2耳聋患者中发现的KCNQ4错义突变在体外培养的细胞中单独表达时表现出功能缺失,且在与野生型共表达时杂合体通道功能减弱,是判断突变是否会引起听力损失的重要依据[2, 3]。
经计算,KCNQ4基因所有可能的错义SNVs总计4,085个,为了前瞻性的检测这些突变体的功能,该研究使用膜片钳技术记录了所有突变体在中华仓鼠卵巢细胞(CHO-K1)中的全细胞电流并进行功能分类。结果显示,70.9% (2,898/4,085)突变体功能正常、26.1% (1,068/4,085)为功能缺失型突变和2.9% (119/4,085)为功能获得型突变。随后模拟DFNA2患者的杂合体基因型状态,将突变型与野生型通道进行1:1共表达,结果表明,516个突变体与野生型共表达后杂合体通道电流依然降低,说明这些突变对野生型KCNQ4介导的电流产生不同程度的抑制作用,可能是由于显性负效应或单倍剂量不足等原因。进一步构建Kcnq4点突变小鼠验证在异源表达系统中检测的杂合体通道功能与在体听力表型的关系,结果表明体外功能分类与Kcnq4点突变小鼠的听力表型高度相符。此外,该研究的功能分类结果与临床致病性解释结果高度一致,可以作为ACMG标准中的强的功能证据BS3和PS3,支持对已发现的KCNQ4 VUS的致病性解析和新发现的错义突变即发现即诊断,对临床诊断和干预起重要参考价值。
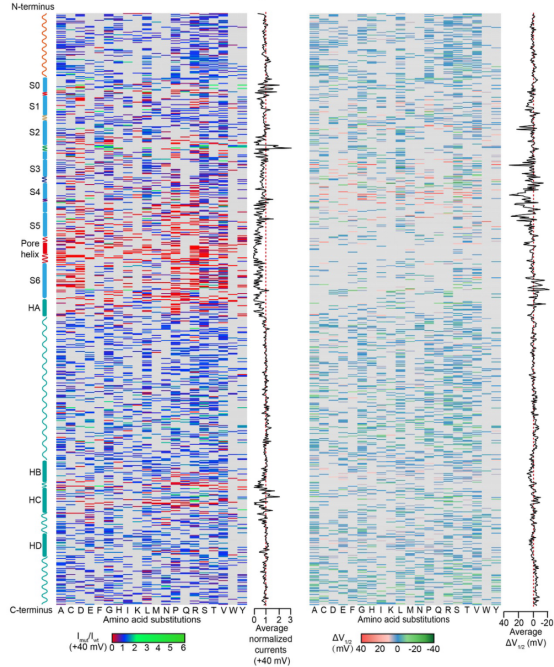
图:KCNQ4所有4,085个错义SNVs单独表达时峰值电流和半激活电压性质。位于S5-Pore-S6结构域的突变多为功能缺失突变。位于跨膜区S1-S4结构域的突变的半激活电压变化较大。灰色表示无相应突变。
综上所述,本研究对遗传性耳聋基因KCNQ4错义突变的功能分析对了解KCNQ4突变的致病机理和DFNA2的诊断有重要参考作用,对遗传性听力损失的临床实践有所帮助。其他离子通道基因,比如同一家族的KCNQ1和KCNQ2,其突变分别可引起遗传性心脏病和癫痫,该文章对KCNQ4的工作展示了对KCNQ家族或其他临床疾病相关电压门控离子通道的所有潜在错义突变进行功能分类的可能性。
复旦大学生命科学学院博士生郑红兰和硕士生严昕昊为本文共同第一作者,深圳湾实验室分子生理学研究所资深研究员闫致强为本文通讯作者。复旦大学金力教授,四川大学华西医院罕见病研究院袁慧军教授和临床遗传学部负责人卢宇主治医师,复旦大学生命科学学院李冠鸾、林恒威、庄文卉、姚富强和夏昕,深圳湾实验室分子生理学研究所邓锶绮也参与了该项研究的部分工作。该研究得到科技部科技创新 2030 —“脑科学与类脑研究”重大项目、科技部国家重点研发计划、国家自然科学基金、上海市科技重大项目、深圳市优秀科技创新人才培养项目、遗传工程国家重点实验室、医学神经生物学国家重点实验室、复旦大学和深圳湾实验室等方面的资助和支持。
参考文献
[1] Cutting, G.R., Annotating DNA Variants Is the Next Major Goal for Human Genetics. American Journal of Human Genetics, 2014. 94(1): p. 5-10.
[2] Kubisch, C., et al., KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell, 1999. 96(3): p. 437-446.
[3] Kim, H.J., et al., Cellular and molecular mechanisms of autosomal dominant form of progressive hearing loss, DFNA2. J Biol Chem, 2011. 286(2): p. 1517-1527.
文章链接:https://doi.org/10.1101/gr.276562.122