饶毅实验室研究进展
从化学分析和生物化学到电生理的多学科途径提示肌酸为新的神经递质
卞希玲、祝婕敏、贾晓波为共同第一作者,梁文君、余思涵、李志强、张文霞为共同作者,饶毅为通讯作者。
神经化学生物学实验室
北京大学 (北大IDG/麦戈文脑研究所、北大清华生命科学联合中心、生命科学学院、化学与分子工程学院化学生物学系、医学部药学院分子与细胞药理学系),北京脑科学研究所,首都医科大学/首都医学科学创新中心,昌平实验室,深圳湾实验室分子生理学研究所,中国医学科学院医学神经生物学研究单元
摘要
发现神经递质,特别是中枢神经系统的,既重要、又困难。我们十二年来寻找新的神经递质。我们在突触囊泡(SV)中检测到肌酸 (Cr),水平低于谷氨酸(Glu) 和伽马氨基丁酸 (GABA)但高于乙酰胆碱 (ACh) 和五羟色胺 (5-HT)、多巴胺(DA)。如果剔除小鼠合成肌酸的酶之一精氨酸:甘氨酸眯基转移酶(AGAT) 的编码基因、或剔除编码SLC6A8的基因(在这一肌酐转运蛋白基因的突变是男性智障最常见的突变) ,都可以显著降低SV内的肌酸。在刺激脑片后,可以检测到肌酸释放。Slc6a8和Agat 突变小鼠的肌酸释放减少。肌酐抑制新皮层椎体细胞。肌酸转运进入突触小体需要SLC6A8。我们发现SV摄入肌酸依赖ATP。我们的生物化学、化学、遗传和电生理结果都与肌酸是神经递质的可能性一致,虽然未达到目前经典神经递质的证明程度。我们发现神经递质的途径是先分析SV中内涵,再确定其功能和生理。
引言
神经信号传递取决于神经细胞与其靶细胞之间的化学传递1-6。神经传递依赖于神经递质、神经调质和神经肽等化学分子。确立一个分子为经典神经递质需要几十年,有时道路曲折7。胆碱能神经信号最初的线索在19世纪就有8-11。胆碱12,13 和乙酰胆碱 (ACh) 14分子找到了几十年后到二十世纪初才知道其药理作用15-17。Henry Dale 及其同事意识到ACh 作用与副交感神经刺激的相似5,18 但直到 1929 才在体内发现ACh 19,到1934 年才证明ACh 是外周神经递质(PNS) 20-22。 而用了一百多年才从发现肾上腺损害23或切除24的影响,观察到肾上腺的活性25,分离其无活性衍生物26-29,成功地分离肾上腺素30-32,注意到肾上腺素与交感神经刺激的类似3,4,33-35,到1940年代中期Ulf von Euler 证明去甲肾上腺素 (NA) 为交感神经递质 36-38。确立外周神经系统的神经递质不容易,确立中枢神经系统的神经递质就更困难。确立ACh为外周递质之后三十多年才确立其为中枢神经递质39-41,确立NA为外周递质二十多年之后才确立其为中枢递质42,43。
如果神经递质只在中枢起作用,而在外周不起作用,要发现或证明这种递质困难大很多。大多数神经递质是按其对外周组织的作用而发现的,以肌肉收缩舒张为主要检出方法。谷氨酸 (Glu) 44-47 和伽马氨基丁酸 (GABA) 46,48-51 的发现部分是由于他们的外周作用、部分是由于他们对脊髓神经元的作用。没有理由中枢递质必须还在外周起作用。但相对很少努力用于发现只在中枢神经系统起作用、而没有外周组织检定方法的小分子神经递质。不成熟的猜测和技术困难是近三十年寻找神经递质不是积极研究领域的主要原因。
有没有更多的神经递质、能否发现它们? 经典神经递质存储于突触囊泡(SVs) 52-60。电刺激导致其释放,之后要么被酶降解、要么被细胞膜的转运蛋白摄入突触前末梢、被囊泡转运蛋白摄入SV 61-66。大多数主要教科书列举三67-69 或 4 70-72 个神经递质的标准:存在于突触前神经元中, 刺激后释放,作用与突触后神经元,移除机理。有些被通常接受为神经递质的分子病不符合不同教科书所列全部标准,但它们在中枢神经系统起重要的功能作用、其缺陷导致人类疾病。在长时间里,不同的小分子被提议为神经递质(例如,教科书46,73),但它们都不符合全部标准。候选分子能否被可靠地在SV中检测到,常常、虽然不是所有时候,是问题所在(参见所引74)。
从2011年开始,我们积极寻找哺乳动物新的神经递质。我们试过不同的途径,包括在脑脊液(CSF)中寻找神经活性物质,追踪可能位于SV的转运蛋白。我们现在走完全的途径是从小鼠脑分离纯化SV,之后化学分析其内涵。我们发现了已知地址如Glu,、GABA、ACh和五羟色胺(5-HT)。但更重要的是,我们在SV中可重复地发现肌酸(Cr) 。
肌酸于1832年被Michel-Eugène Chevreul所发现 75,76,长期被认为是肌肉和脑中的能量缓冲77-79。哺乳动物一半的肌酸被认为来自饮食、其余来自内源合成80。多数肌酸存在于肌肉,但也有部分在脑中。虽然内源合成多在肾脏、胰腺和肝脏77,81,但也有脑内合成的肌酐80,82,83。
SLC是一个大的转运蛋白家族,有些跨细胞膜转运、有些跨囊泡膜转运。SLC6亚家族在人类有19个成员,依赖Na+ 或H+ 的梯度进行主动转运84-89。SLC6也被称为神经递质转运蛋白 (NTT)家系,因为其中有些成员转运神经递质如GABA (由 SLC6A1或GABA 转运蛋白1, GAT1; SLC6A13或GAT2; SLC6A11或GAT3) 90-93,NA (由SLC6A2或NA转运蛋白,NET) 65, 多巴胺 (由SLC6A3或DAT) 94-96,5-HT (由SLC6A4或五羟色胺转运蛋白,SERT) 66,97,和甘氨酸 (由 SLC6A9, 或GlyT1;SLC6A5或GlyT2) 90,98,99。肌酐由SLC6A8转运 (亦称肌酐转运蛋白CrT, CT1或CRCT) 100-106。除了外周组织和器官, SLC6A8也表达于神经系统,主要是神经元中82,104,107-109。SLC6A8 蛋白质位于神经元细胞膜上109-111。
SLC6A8在脑内的功能重要性由Slc6a8基因有缺陷的人所显示的症状所支持。Slc6a8 基因的突变见于有智障(ID)的病人,语言发育延缓、癫痫发作、自闭症样行为112,113。这类疾病都归为肌酐转运缺陷 (CTD),以ID 为显著特征。有些Slc6a8 基因突变的病人显示语言发育的特别敏感性,他们智障轻微而语言延缓严重114。CTD占X染色体相关的智力滞后的1 到2.1% 115-123。虽然CTD在智障男性特别高,也有女性CTD,大约携带率为0.024% 124。
Slc6a8 基因敲除小鼠125-127 显示人类CTD病人的典型症状:早发、进行性学习记忆损害。在脑或在神经元特异敲除Slc6a8基因的小鼠也显示学习记忆缺陷,而没有Slc6a8在外周作用所导致的运动变化128,129。在脑内多巴胺神经元特异敲除Slc6a8 导致运动过度130。 这些结果表明SLC6A8 在神经元内发挥重要功能。
肌酸缺乏综合症(CDS)是肌酐代谢先天疾病,可以有以下三个基因之一缺陷所致:胍乙酸甲基转移酶(Gamt)131,精氨酸-甘氨酸眯基转移酶(Agat) 132,133, and Slc6a8 112。它们都有脑疾病说明脑内肌酐的功能重要性134-137。
在此我们先从小鼠脑经过生物化学分离纯化SV,发现SV含肌酸,以及经典神经递质如Glu、GABA、ACh和5-HT。 然后我们检测在升高细胞外钾离子(K+)而使神经元去极化的时候,钙离子(Ca2+)依赖的肌酐、Glu和GABA释放,而没有检测到ACh和5-HT的释放。遗传敲除Slc6a8基因或Agat基因都可以降低SV含肌酐的水平、降低刺激引起的肌酐释放。当把肌酐加到新皮层的脑片时,锥体细胞的活动被抑制。我们进一步验证肌酐可以被突触小体所摄取,并发现敲除Slc6a8基因可以显著减少突触小体摄取肌酐。最后,我们发现肌酐可以被转运到SV中。因此,综合化学分析、生物化学、遗传学和电生理的多学科研究提示肌酐是新的神经递质,可以有找到肌酐受体来进一步证明。
结果
小鼠脑内突触囊泡内检测到肌酸
为了寻找新的神经递质,我们试了几个途径。例如,我们用钙离子(Ca2+)成像检测脑脊液(CSF)中神经活性物质,但很难去除已知神经递质、选择潜在的新递质的反应。我们也把全部人类SLC的cDNA转染到小鼠脑分离到的原代神经元中,发现多于50个SLCs可以表达在SV。但是,当我们用CRISPR-Cas9标记小鼠一些候选SLC时,发现其中有些在中枢神经系统之外表达,而只是异位表达时才出现在SV上,内源的不在SV上。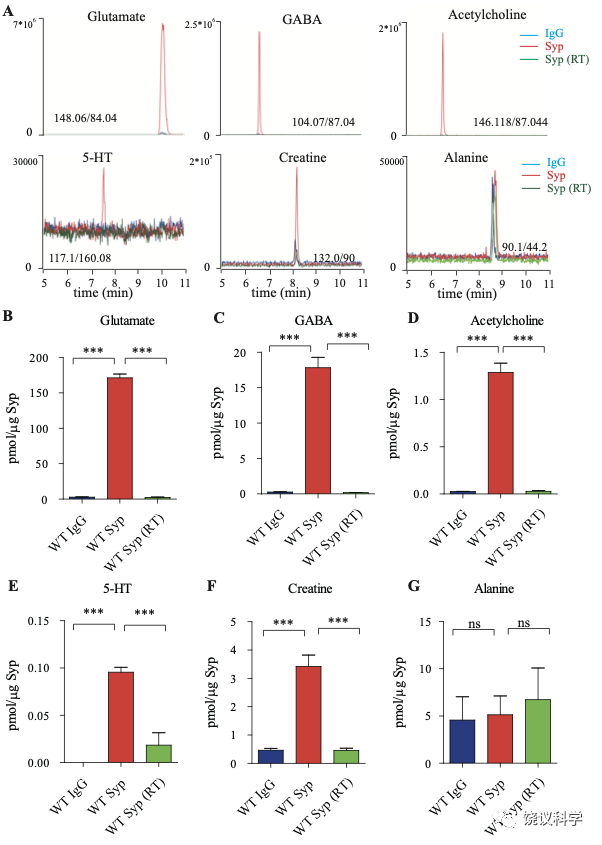
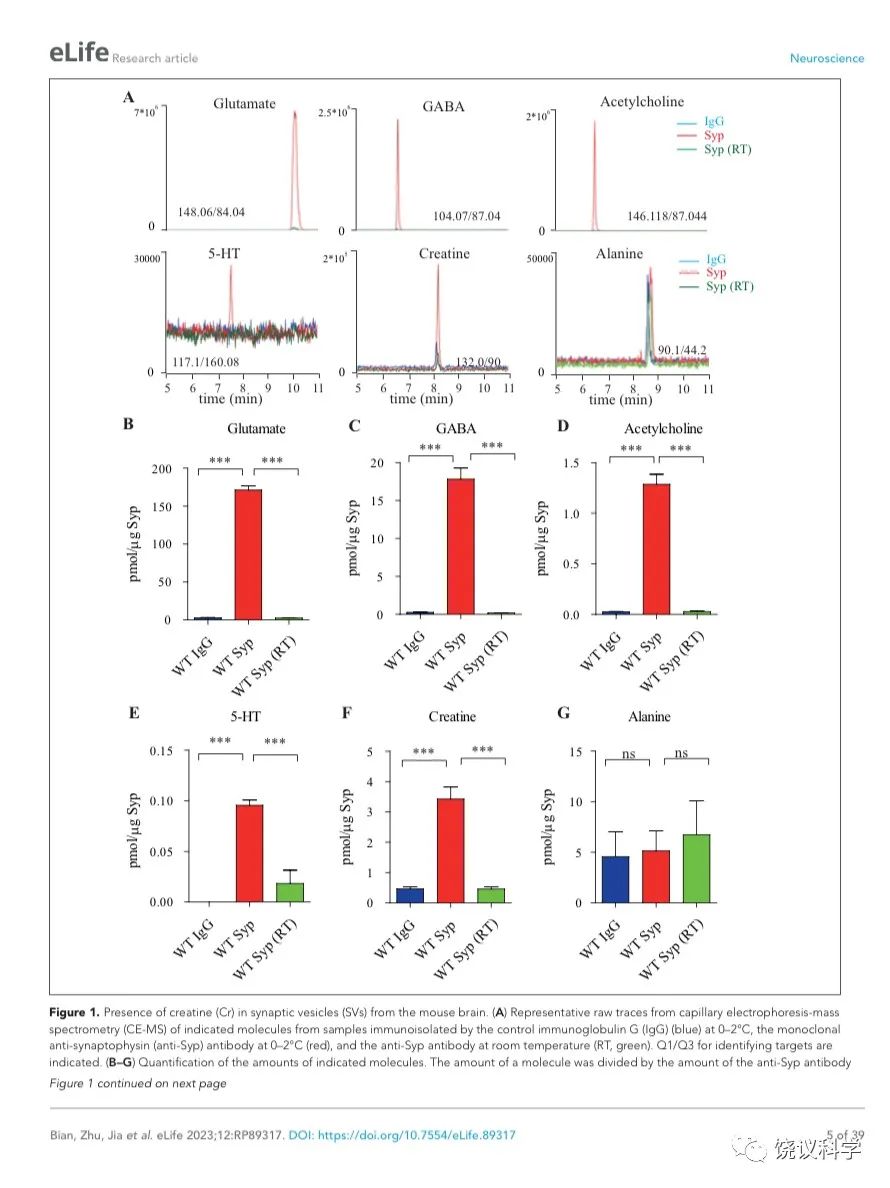
图1. 小鼠脑的SV存在肌酐。(A) Representative raw traces from CE-MS of indicated molecules from samples immunoisolated by the control IgG (blue) at 0-2 ºC, the monoclonal anti-Syp antibody at 0-2 ºC (red) and the anti-Syp antibody at room temperature (RT, green). Q1/Q3 for identifying targets were indicated. (B-G) Quantification of the amounts of indicated molecules. The amount of a molecule was divided by the amount of the anti-Syp antibody bound to magnetic beads. Note Glu (B), GABA(C), ACh (D), 5-HT (E), Cr (F), but not alanine (G) was higher in SVs pulled down by the anti-Syp antibody at 0-2 oC than those pulled down by the IgG control or those pulled down at the RT. n=10 (B-E, and G) or 14 (F) samples per group, ***, p<0.001, ns, not significant. one-way ANOVA with Tukey’s correction.
在此,我们用的途径是以纯化SV作为第一步(图1—附图 1A) 138-145。 Synaptophysin (Syp) 是SV的特异标记146-149,抗Syp抗体可以用于免疫分离SV143,144,150-152。电子显微镜(EM)监测(图1—附图 1B左)显示纯化的囊泡均质性,平均直径40.44 ± 0.26 纳米 (数了596 个颗粒) (图1—附图1B, 右),与以前对SV的描述一致153,154。
免疫印迹分析了神经元的二十个亚细胞标记物和神经胶质细胞的一个标记物 (图1—附图1C)显示我们的纯化高度有效,用抗-Syp抗体纯化后可以检测到SV的标记物,用对照免疫球蛋白(IgG)纯化的不行。SV蛋白包括Syp 146-149, synaptotagmin (Syt1) 155,synatobrevin2 (Syb2) 156,157, SV2A 158, H+-ATPase 159, 以及囊泡的突触转运蛋白如谷氨酸转运蛋白(VGLUT1, VGLUT2) 160和GABA 转运蛋白(VGAT) 161。抗-Syp免疫分离没有带下以下标记物:突触膜 (以SNAP23 为标记物) 162-164,突触后成分 (以PSD95和GluN1为标记) 165,166,高尔基体(以GM130和Golgin 97为标记) 167,168,早期内吞体(以EEA1为标记) 169,溶酶体 (以LC3B和组织蛋白酶B为标记), 细胞浆(以3-磷酸甘油醛脱氢酶GAPDH为标记), 线粒体(以电压依赖阴离子通道VDAC为标记),细胞膜(以电压门控钙离子通道alpha 1亚基CACNA1A为标记),轴突膜(以4型葡萄糖转运蛋白GluT4为标记) 和神经胶质细胞膜(以髓磷脂碱性蛋白MBP为标记)。这些结果表明我们获得的SV高纯度完好。
为了检测和定量纯化的SV中缩合的候选递质小分子,使用和优化了毛细管电泳-质谱(CE-MS) (图 1A, 附图 1A) 151,170。我们发现经典神经递质如Glu、 GABA、ACh和5-HT抗Syp抗体拉下的SV中水平显著高于对照IgG拉下的(图1 A-E)。与以前报道一致144,151,只在摄氏0度,而不能在室温(RT)观察到免疫分离的SV中神经递质的富集(图1 A-E)。而另外的小分子丙氨酸(图1G),其SV水平与对照没有提高。
Glu水平为 171.1 ± 5.4 pmol/μg anti-Syp antibody(皮摩尔/微克抗Syp抗体) (10次,图1B),约十倍于GABA (n=10,17.81 ± 1.47,皮摩尔/微克抗Syp抗体, 图1C)。ACh量为1.29 ± 0.10 皮摩尔/微克抗Syp抗体(n=10, 图1D),约为GABA的0.072。5-HT量为 0.096 ± 0.017 皮摩尔/微克抗Syp抗体 (n=10, 图1E)。因此,我们的纯化和检测方法高度可靠、灵敏到可以检测已经确立的神经递质。
在同样情况下,我们可以检测到SV内含肌酐(n=14,图 1A和1F)。 SV内含肌酐量为3.43 ± 0.40皮摩尔/微克抗Syp抗体(图1F),约位Glu的 2% of Glu、 GABA的19%、ACh的266%、5-HT的3573%。这些差别不太可能是因为不同神经递质在单个SV的浓度差别所导致,而更可能是因为含不同神经递质的SV相对丰度的差别。小鼠脑内85%至90%的神经元是谷氨酸能的,而10%至15%为GABA能171-173,这可以接受为什么我们检测到的Glu约位GABA的十倍(图 1B和C)。同样,胆碱能神经元 (5.67x105) 174 占小鼠脑内神经元总数(约7千万)的0.81% 175,五羟色胺能神经元(约 2万6千个) 占全部神经元的0.037% 175,176。 假定在单个SV中每一种神经递质的含量相似,从以上资料可以推测小鼠脑中约1.3% 到2.15% 的神经元为肌酐能。
为了区分与SV共分离的小分子是在囊泡内143,144,还是附在囊泡外154,我们检测了SV中这些分子存在的对稳定和氢离子(H+)电化学梯度的依赖性。室温纯化的SV含肌酐量显著低于0 ºC免疫分离的 (图 1F),支持肌酐是在SV内,而不是其外。
经典神经递质存储在内部酸性环境的SV中(pH of 5.6 ~ 6.4) 177-179。 为验证肌酐储存于SV内、检测H+电化学梯度的作用,我们在纯化SV的过程中用了药理抑制剂74,180。用质子载体FCCP (carbonyl cyanide-4-(tri-fluoromethoxy) phenylhydrazone)去除H+电化学梯度180, 181。 FCCP 显著减少SV的肌酐和经典神经递质含量(图1—附图2A-E)。FCCP诱导的下降程度与不同分子的pKa 或PI (等电点)相关:5-HT (预计pKa 为 10 和9.31, 图1—附图2E) > Cr (PI约 ~7.94,图1—附图 2A) > GABA (PI 为7.33,图1—附图 2C) > Glu (PI为3.22,图1—附图2B)。可以驱散ΔpH 的K+/H+ 离子交换剂Nigericin 180,181也减少SV的肌酐和经典神经递质含量 (图 1—附图2A-E)。而且,在FCCP或nigericin存在的情况下,SV的肌酐降低到与对照IgG拉下的肌酐水平一样(图1—附图 2A), 显示SV储存肌酐是依赖于H+ 梯度的。作为对照,这些抑制剂并不改变SV非神经递质分子如丙氨酸的水平(图1—附图2F)。
缺乏Slc6a8基因小鼠的SV含肌酸下降
位于X染色体的Slc6A8编码肌酐的转运蛋白,其缺乏功能 (LOF) 的突变在人112,113和小鼠125-130都导致行为的缺陷。为研究SLC6A8是否影响SV的肌酐,我们制造了Slc6a8基因敲除的小鼠。用CRISPR/Cas9基因修饰体系,用CreERT2-WPRE-polyA 替代了Slc6a8基因外显子1的一部分(图2A)。反向转录-多聚酶链反应(RT-PCR) (图2—图1A和B)以及定量真时反向PCR (qPCR,图 2—附图1C和D) 显示在雌性或雄性突变小鼠中不能检测到Slc6a8的mRNA,而在雌性杂合鼠(Slc6a8+/-)中其含量也显著降低。与以前的报道一致,Slc6a8 KO(基因敲除)小鼠的体重减低 (图2—附图2B 和D) 125,182,183。 Slc6a8 KO 与野生型(WT)小鼠的脑中没有显著差别(图2—图2A和C)。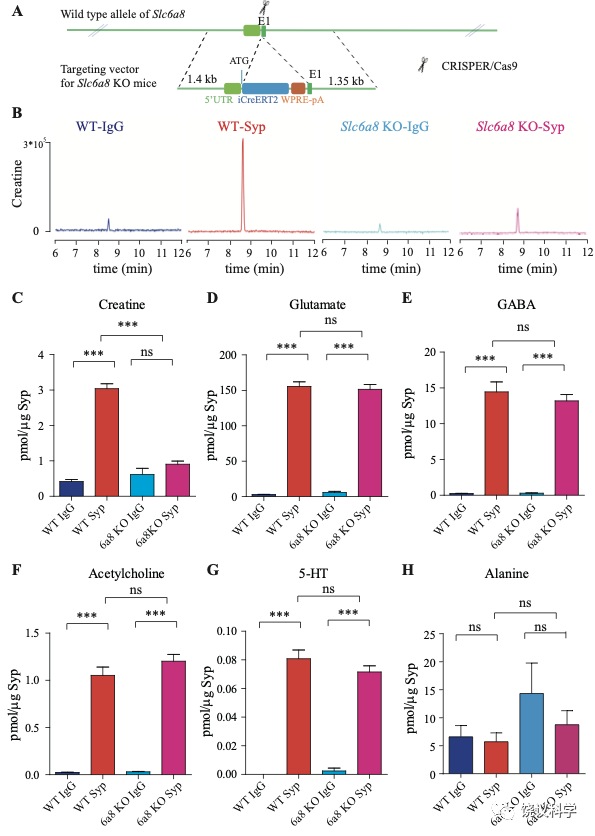
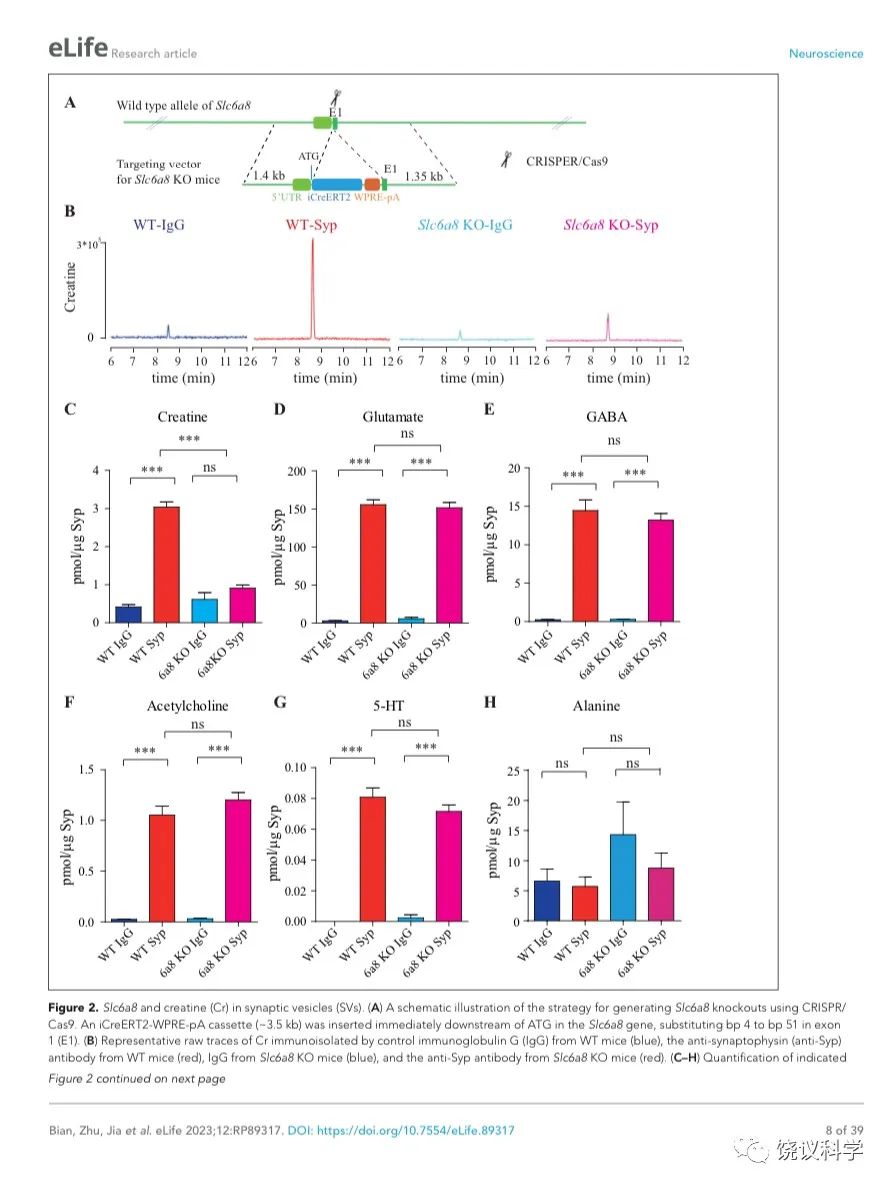
图2. SV含Slc6a8和肌酐Cr in SVs. (A) A schematic illustration of the strategy for generating Slc6a8 knockouts using CRISPR/Cas9. An iCreERT2-WPRE-pA cassette (~3.5 kb) was inserted immediately downstream of ATG in the Slc6a8gene, substituting bp 4 to bp 51 in exon 1 (E1). (B) Representative raw traces of Creatine immunoisolated by control IgG from WT mice (blue), the anti-Syp antibody from WT mice (red), IgG from Slc6a8 KO mice (blue) and the anti-Syp antibody from Slc6a8 KO mice (red). (C-H) Quantification of indicated molecules. Note the selective reduction of Cr in SVs from Slc6a8 KO mice. n=14 samples per group. ***, p<0.001, ns, not significant. one-way ANOVA with Tukey’s correction.
当我们检测抗-Syp抗体对比Igg拉下的SV的内含,只有肌酐含量下降,而经典神经递质的含量没有下降(图2 B-H, 图2—附图3A-E)。虽然IgG拉下的肌酐含量在Slc6a8-/Y 与Slc6a8+/Y 小鼠之间没有显著差别,抗-Syp抗体从,Slc6a8-/Y 小鼠拉下的SV肌酐含量约为同窝WT (Slc6a8+/Y) 小鼠脑拉下的三分之一(n =14, 图2B-C)。与IgG对照相比,WT小鼠SV富集肌酐,而Slc6a8-/Y小鼠的SV不富集肌酐(图2 B 和C)。在Slc6a8-/Y和Slc6a8+/Y小鼠,经典神经递质都在SV中富集(相比于IgG对照) (图2D-G,图2—附图3A-D)。SV含Glu (图2D和图2—附图3A)、GABA (图2E和图2—附图3B)、ACh (图2F 和图2—附图3C)、5-HT (图2G和图2—图3D) 的量在Slc6a8-/Y小鼠与Slc6a8+/Y小鼠之间无显著差异。WT小鼠的SV不富集的分子如丙氨酸,也不受Slc6a8 KO所影响(图2H和图2—附图3E)。
Slc6a8 KO小鼠中分离的SV所含肌酐的特异下降,不太可能是由于技术伪迹。首先,从Slc6a8 KO小鼠获得的SV量减少的可能性被免疫印迹分析所排除,因为SV标记物如Syp、Syt及H+-ATPase都没有降低(图2—附图4A)。其次,用峰面积定量高分辨率质谱(Q Exactive HF-X)收集的资料也揭示Slc6a8 KO小鼠选择性降低SV中肌酐(m/z=132.0576)含量(n=8,图2—附图4B-G)。而Glu (n=8, 图2—附图4B, m/z=148.0604)、GABA (n=8, 图2—附图4C, m/z=104.0712)、ACh (n=8, 图2—附图4D, m/z = 146.1178)以及 丙氨酸(n=8, Ala, 图3—附图4E, m/z=90.055)的峰面积在用抗Syp抗体和对照IgG从WT和Slc6a8 KO小鼠拉下的SV之间都没有显著差别。但是,从WT小鼠拉下的SV之肌酐的峰面积(图2—附图4G)与肌酐幅度(n=8, 图2—附图4F) 信号都显著高于IgG拉下的,而从Slc6a8 KO小鼠拉下的却无差别。
缺乏Agat基因小鼠的SV含肌酸下降
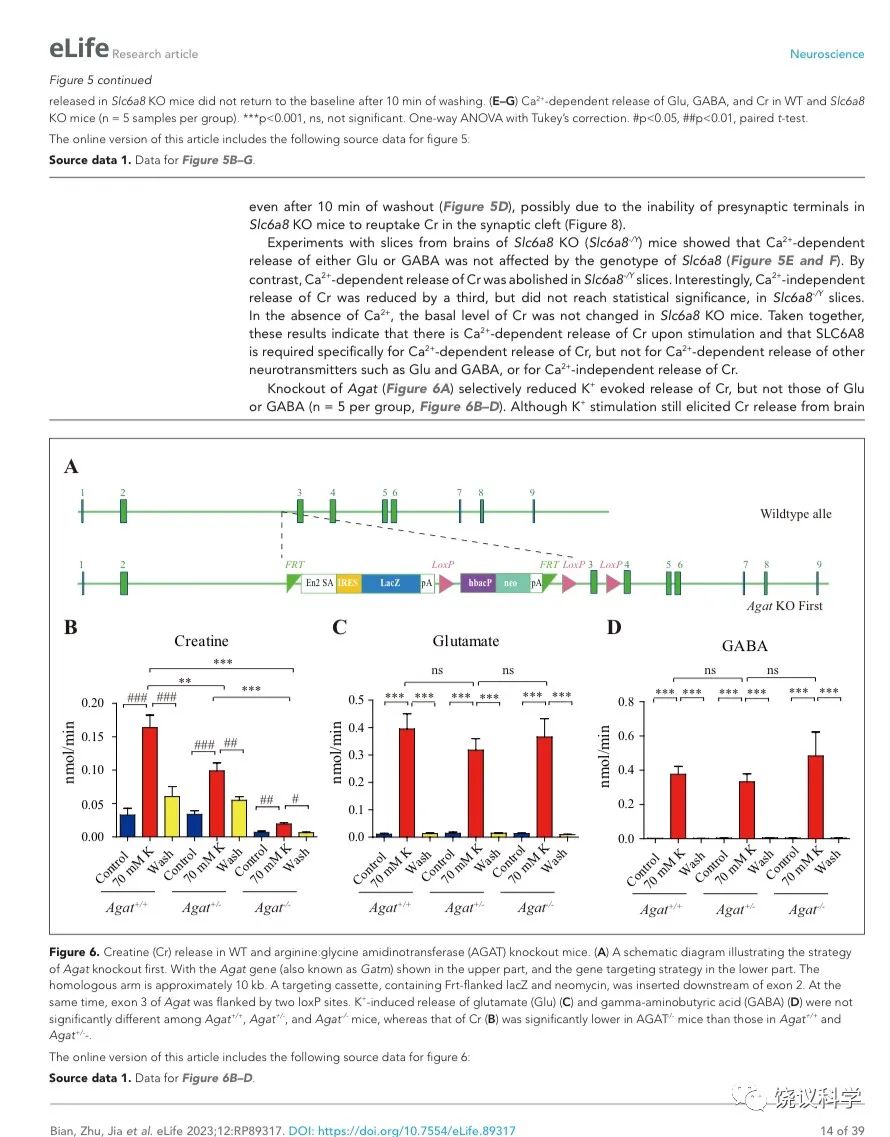
AGAT是催化肌酐合成第一步的酶82,184,其缺陷也导致人脑Cr缺乏及智力滞后132,133。
为研究AGAT对于SV的肌酐是否必需,我们用了Agat 基因“knockout-first” 的小鼠(图6A) 185。靶向盒含Frt (Flip重组位点)-包围的EnS2A、IRES::lacZ陷阱盒, flox位点包围的 neo 基因盒被插到第二个外显子的下游以干扰Agat pre-mRNA的正常剪接。RT-PCR (图3—附图 1A)与定量RT-PCR (图3—附图1B) 检测显示Agat+/- 小鼠的Agat mRNA 含量降低、而在Agat-/-小鼠中检测不到Agat mRNA。Agat-/-小鼠的体重(图3—附图2B)低于Agat+/+以及Agat+/-小鼠,但脑重(图3—附图2A)不低,这些类似Slc6a8 KO 小鼠。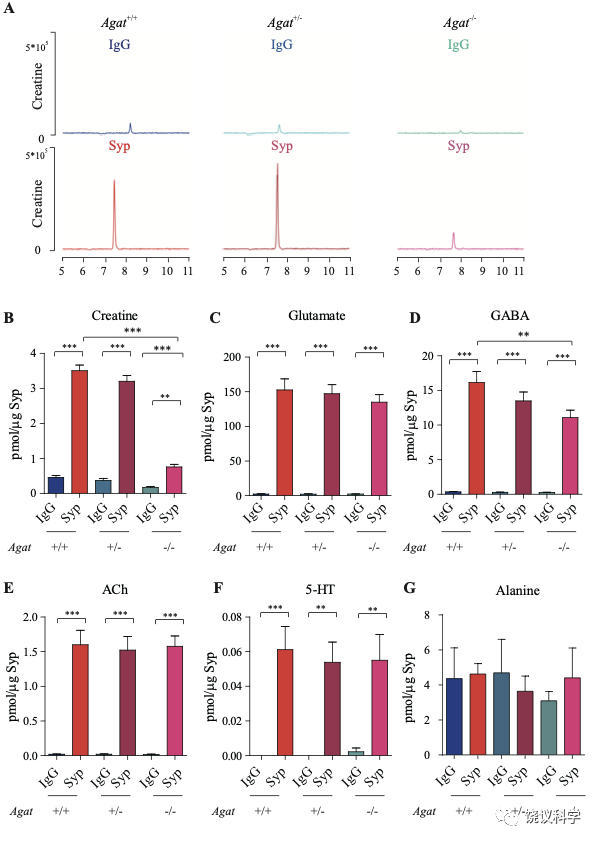
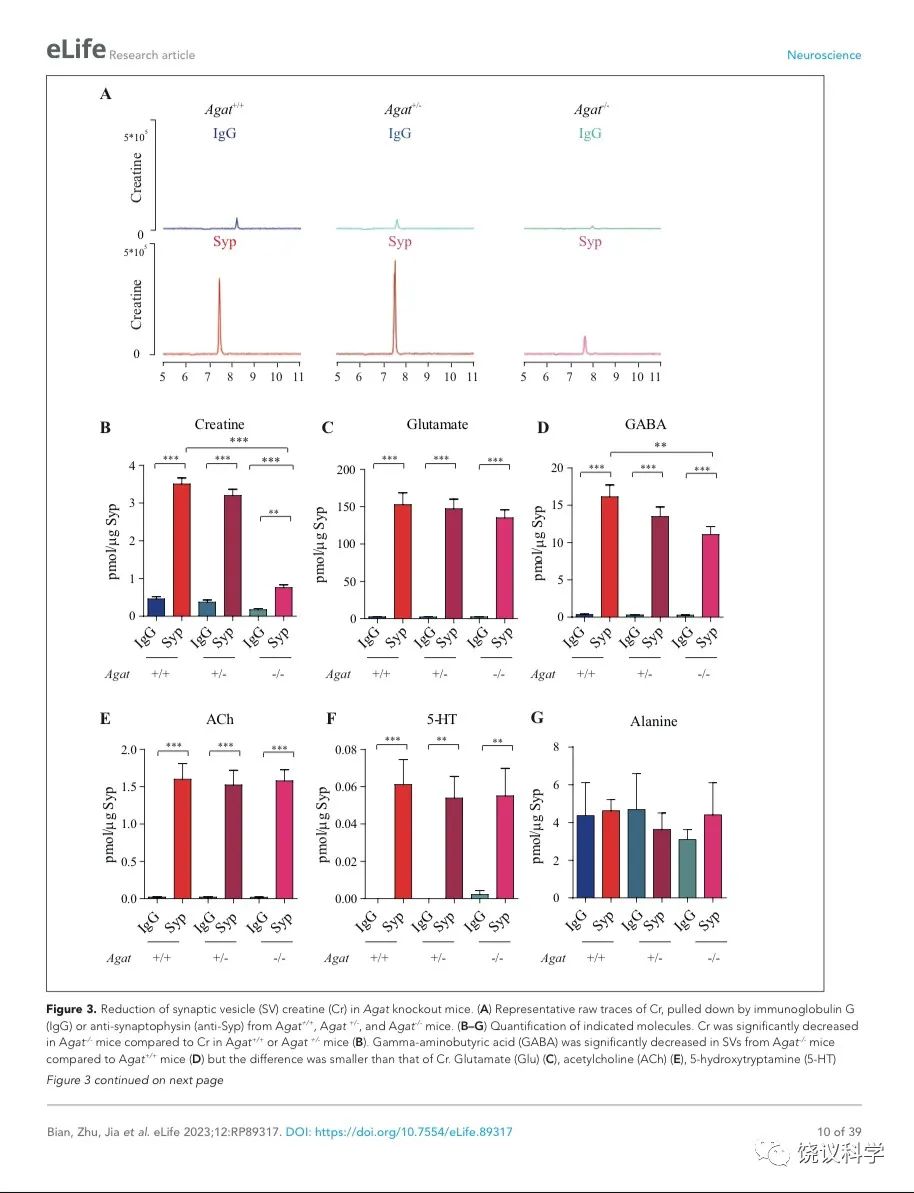
图3. Reduction of SV Cr in Agat基因剔除小鼠. (A) Representative raw traces of Creatine, pulled down by IgG or anti-Syp from Agat+/+, Agat+/- and Agat-/- mice. (B-G) Quantification of indicated molecules. Cr was significantly decreased in Agat-/- mice as compared to Cr in Agat+/+ or Agat+/-mice (B). GABA was significantly decreased in SVs from Agat-/-mice as compared to Agat+/+ mice (D) but the difference was smaller than that of Cr. Glu (C), ACh (E), 5-HT (F) and alanine was not different among Agat+/+, Agat+/- and Agat-/-mice. n=10 samples per group. ***, p<0.001, ns, not significant. one-way ANOVA with Tukey’s correction.
由定量分析Syp、Syt和H+-ATPase所支持 (n=20,10个样本含两次重复),免疫印迹分析显示Agat+/+、Agat+/-和Agat-/-三种基因型小鼠脑内分离到的SV没有显著差异(图3—附图3A-D)。
我们分析从Agat+/+、Agat+/-和Agat-/-三种基因型小鼠脑分离到的SV里面所含小分子。三种基因型得到的SV中肌酐含量都显著高于IgG对照(图3A和3B)。但是,脑SV 内含肌酐水平在Agat-/-小鼠显著低于Agat+/+与Agat+/-小鼠 (n=10, 图3A和B)。
Glu (图3C)、ACh (图3E)和5-HT (图3F)皆在SV中富集(相比于IgG对照),而且在Agat+/+、Agat+/-和Agat-/-三种基因型小鼠脑无显著差别。GABA in SVs from Agat-/-小鼠脑SV的GABA低于Agat+/+小鼠约30%,幅度不如肌酐(78.4%)。丙氨酸在三种基因型小鼠的SV中没有差别(n=6,图3G)。所以,Agat KO 小鼠的Cr和GABA下降,而其他神经递质没有下降。
基因敲入小鼠所显示的SLC6A8基因表达范式
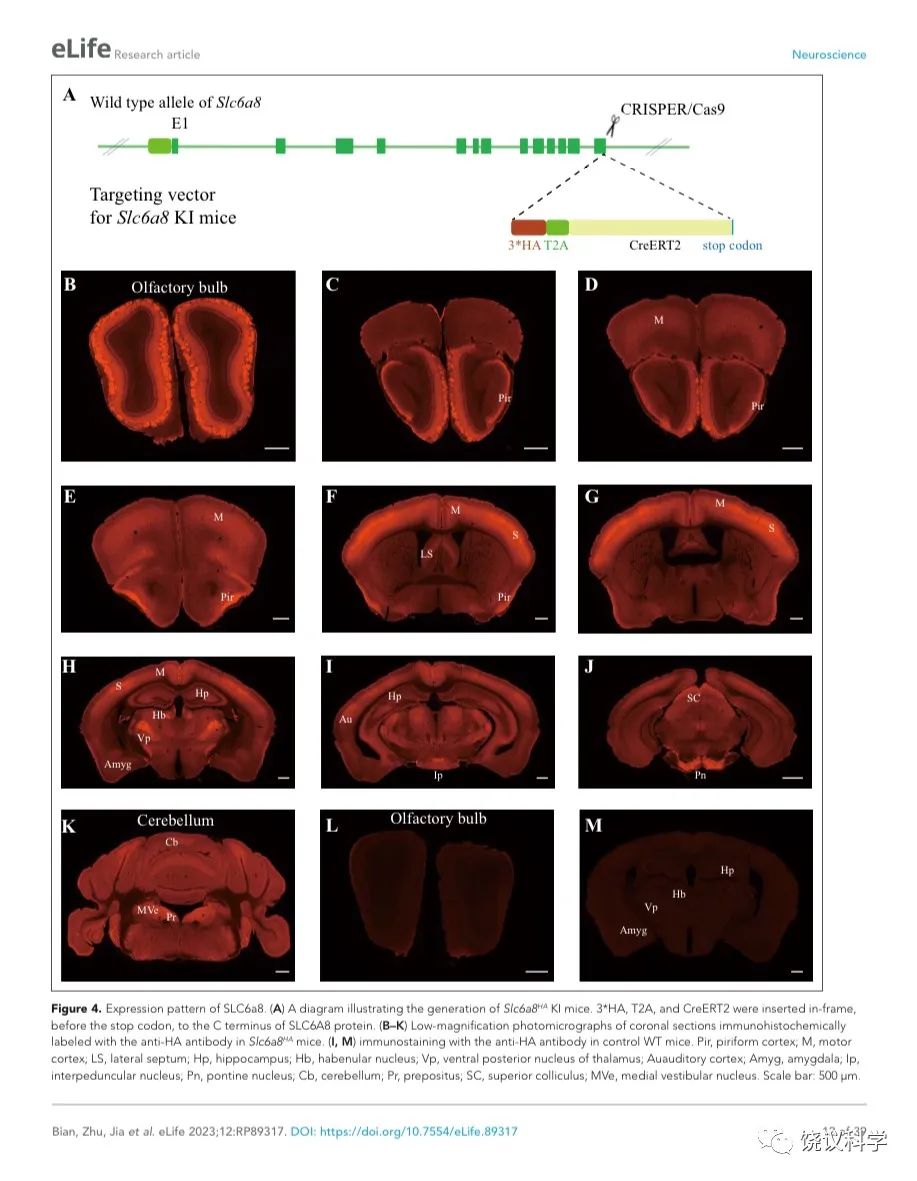
我们用CRISPR/Cas9制造了Slc6a8HA基因敲入小鼠。在SLC6A8蛋白质的C末端同框插入了三重血凝素标签(HA) 186、T2A序列187-189和CreERT2 190-193 (图4A)。
为检测SLC6A8表达范式,我们在Slc6a8HA和WT小鼠用抗HA抗原镞的抗体进行免疫细胞化学染色。Slc6a8HA小鼠显示以下部位有阳性信号:嗅球(图4B)、梨状皮层(图4 C-F)、体躯感觉皮层(图4F和G)、腹后丘脑 (图4H)、脚间核(图 4I)和脑桥核(图4J)。此外,中等水平的免疫反应性还见于:运动皮层 (图4 D-H)、内侧缰核(图4H)、海马(图4H)和小脑(图4K)。这些结果与以前报告一致 109,111。 WT小鼠在抗HA抗体染色时阴性(图4 L和M)。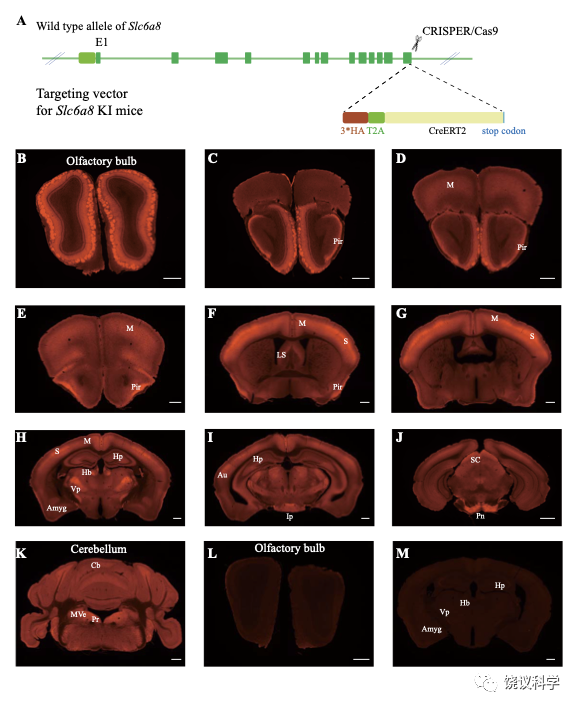
图4. SLC6a8表达模式. (A) A diagram illustrating the generation of Slc6a8HA KI mice. 3*HA, T2A and CreERT2 were inserted in-frame, before the stop codon, to the C terminus of SLC6A8 protein. (B-K) Low magnification photomicrographs of coronal sections immunohistochemically labelled with the anti-HA antibody in Slc6a8HA mice. (I and M) immunostaining with the anti-HA antibody in control WT mice. Abbreviations: Pir, piriform cortex; M, motor cortex; LS: lateral septum; Hp: hippocampus; Hb, habenular nucleus; Vp: ventral posterior nucleus of thalamus; Au auditory cortex; Amyg: amygdala; Ip, interpeduncular nucleus; Pn; pontine nucleus; Cb, cerebellum; Pr, prepositus; SC, Superior colliculus; MVe, medial vestibular nucleus. Scale bar: 500 μm.
刺激引起的钙离子依赖的肌酐释放
刺激后,经典神经递质以Ca2+依赖模式释放到突触间隙。例如,高钾(K+)刺激脑片以Ca2+依赖模式释放Glu、GABA和其他神经递质 194-198。
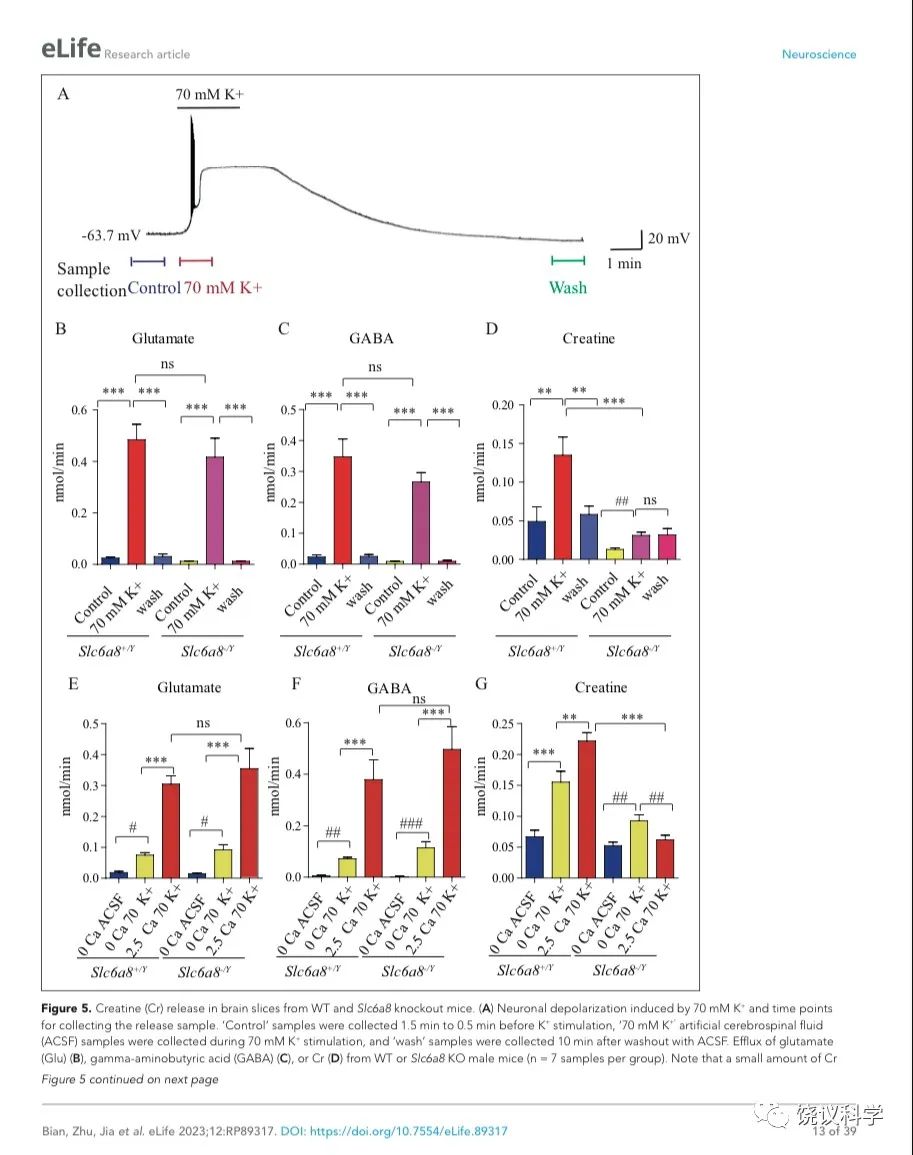
我们用了前囟后1~2毫米(mm)处的冠状脑切片(厚300 μm),因为皮层、丘脑、缰核和海马都呈SLC6A8阳性(参考图4H)。我们通过记录脑片神经元监测钾刺激的作用。在K+刺激后,海马CA1区域的椎体神经元立即去极化,发放一窜动作电位,不到一分钟(min)就达到大的去极化平台期(图5A)。K+刺激诱导的去极化持续几分钟,然后趋于基线,10 mins 可以洗脱。因此,K+刺激前后每1.5 min或洗脱后十分钟分别收集了1-min的流出液 (图5A),其中的代谢物经CE-MS分析。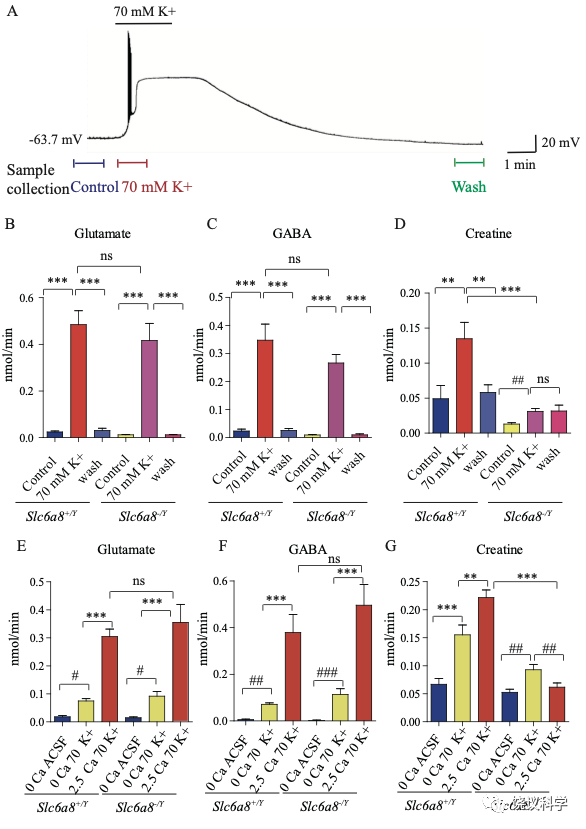
图5. 野生型和Slc6a8基因敲除型小鼠脑片肌酐释放. (A) Neuronal depolarization induced by 70 mM K+ and time points for collecting the release sample. “Control” samples were collected 1.5 min to 0.5 min before K+ stimulation, “70 mM K+” ACSF samples were collected during 70 mM K+ stimulation, and “wash” samples were collected 10 mins after washout with ACSF. Efflux of Glu (B), GABA (C) or Cr (D) from WT or Slc6a8 KO male mice (n=7 samples per group). Note that a small amount of Cr released in Slc6a8 KO mice did not return to the baseline after 10 mins of washing. (E-G) Ca2+ dependent release of Glu, GABA and Cr in WT and Slc6a8 KO mice (n=5 samples per group). ***, p<0.001, ns, not significant. one-way ANOVA with Tukey’s correction. #, p<0.05, ##, p<0.01,paired t test.
在Ca2+存在的情况下,提高细胞外K+引起去极化造成来自WT (Slc6a8+/Y)小鼠的脑片释放Glu和GABA (每组n=7,图5 B与C)。洗脱10 mins后, Glu和GABA的水平回归基线 (图5 B和C)。在Ca2+存在的情况下,提高K+引起去极化导致肌酐释放。10 min洗脱后,细胞外肌酐归于基线(图5D)。为了定量,刺激释放的代谢物是从K+刺激引起的总释放减去基线水平而得。在Ca2+存在的情况下,K+刺激引起Glu、GABA和肌酐流出速度分别为0.46、0.33和0.086 nmol/min (每组n=7) (图5 B, C, D)。按我们系统检测ACh 和5-HT的极限,我们推测ACh流出速度低于0.001 nmol/min、5-HT外流速度低于0.003 nmol/min。脑片肌酐的流出速度低于Glu和GABA的,但高于ACh和5-HT的。
递质释放的Ca2+依赖性通过比较无Ca2+ACSF溶液而提高K+ (补1 mM EGTA),缺Ca2+而提高细胞外K+(补1 mM EGTA),或K+ 在2.5 mM Ca2+ (图5 E-G,每组n=5)。无Ca2+时,提高K+刺激引起小量、但显著量的Glu和GABA释放,流出速度分别为0.056和0.066nmol/min (图5 E, F)。有2.5 mM Ca2+时,提高K+进一步5到6倍地提升Glu和GABA释放,验证以前报道的去极化引起Ca2+依赖的神经递质释放194,196.
肌酐也有Ca2+依赖和Ca2+不依赖的释放(图5G)。有2.5 mM Ca2+时,K+刺激引起的肌酐释放多于缺Ca2+时K+刺激引起的肌酐释放。这些结果显示刺激后有钙依赖的肌酐释放。
Slc6a8和Agat基因突变小鼠肌酸释放减少
我们检测Slc6a8 KO是否影响K+引起的肌酐释放。Slc6a8 KO (Slc6a8+/Y) 小鼠脑片释放的Glu和GABA水平不显著低于WT小鼠(图5 B, C),K+刺激Slc6a8-/Y小鼠脑片释放肌酐显著低于Slc6a8+/Y小鼠(图5D)。Slc6a8 KO (Slc6a8+/Y) 小鼠脑肌酐的基础水平就低于WT小鼠。另外,K+刺激Slc6a8 KO小鼠脑片引起的肌酐释放即使是在洗脱十分钟后,还在持续,可能是因为Slc6a8 KO小鼠的突触前膜不能从突触间隙再摄取肌酐(图8)。
用 Slc6a8 KO (Slc6a8-/Y) 小鼠脑片的实验显示Slc6a8的基因型不影响Glu或GABA的释放(图5 E和F)。与之形成对照的是,Slc6a8-/Y脑片完全没有Ca2+ 依赖的肌酐释放。有趣的是,不依赖Ca2+的肌酐释放只在Slc6a8-/Y脑片降低三分之一,但没有达到统计显著性。缺Ca2+时,Slc6a8 KO小鼠不改变肌酐的基线水平。总结起来,这些结果显示刺激时有Ca2+ 依赖的肌酐释放,SLC6A8只特异的为Ca2+ 依赖的肌酐释放所必需,而不为Ca2+ 依赖的其他神经递质如Glu和GABA的释放所必需,也不为Ca2+不依赖的肌酐释放所必需。
剔除Agat基因(图6A)选择性地减少K+引起的肌酐释放,而不影响Glu或GABA (每组n=5,图 B, C, D)。虽然K+刺激还能引起Agat-/-小鼠脑片的肌酐释放,流出速度减到了Agat+/+小鼠脑片的10%以下、Agat+/-小鼠脑片的20%以下。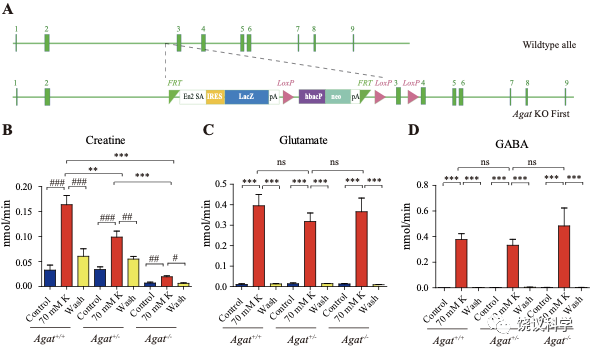
图6. 野生型和Agat基因敲除型小鼠肌酐释放. (A) A schematic diagram illustrating the strategy of Agat knockout first. With the Agat gene (also known as Gatm) shown in the upper part, and the gene targeting strategy in the lower part. The homologous arm is approximately 10 kb. A targeting cassette, containing Frt-flanked lacZ and neomycin, was inserted downstream of exon 2. At the same time, exon 3 of Agat was flanked by two loxP sites. K+ induced release of Glu (C) and GABA (D) were not significantly different among Agat+/+, Agat+/- and Agat-/- mice, whereas that of Cr (B) was significantly lower in AGAT-/- mice than those in Agat+/+ and Agat+/- mice.
肌酸抑制新皮层神经元
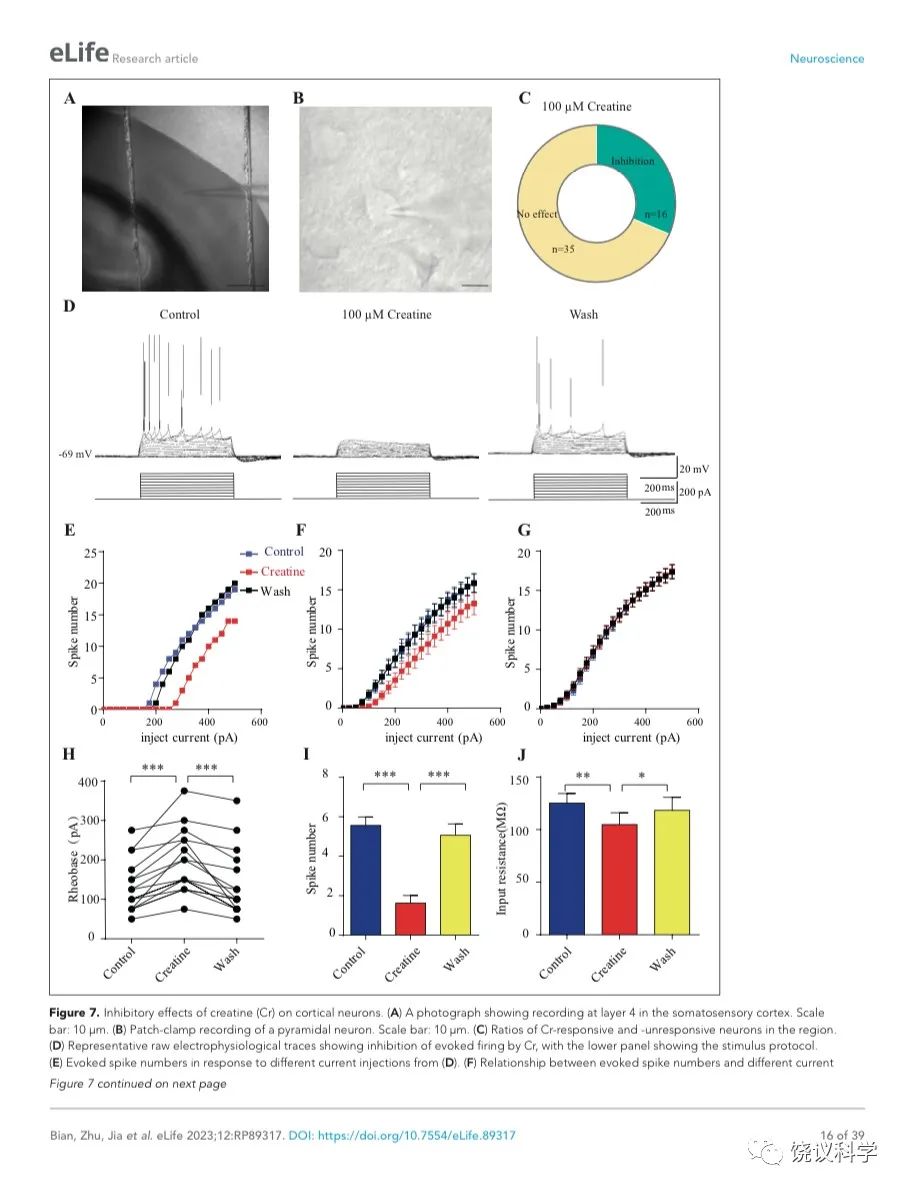
我们自己的资料(图4H)与以前的报道 109,111显示新皮层有SLC6A8,第4层SLA6A8-HA免疫反应神经纤维致密(图7—附图1A) 199,200。体躯感觉皮层的第5层神经元此前就被报道表达SLC6A8 109,111。为研究肌酐的电生理作用,我们用全细胞膜片钳技术记录体躯感觉皮层的第4/5层的椎体细胞 (图7 A和B,图7—附图1)。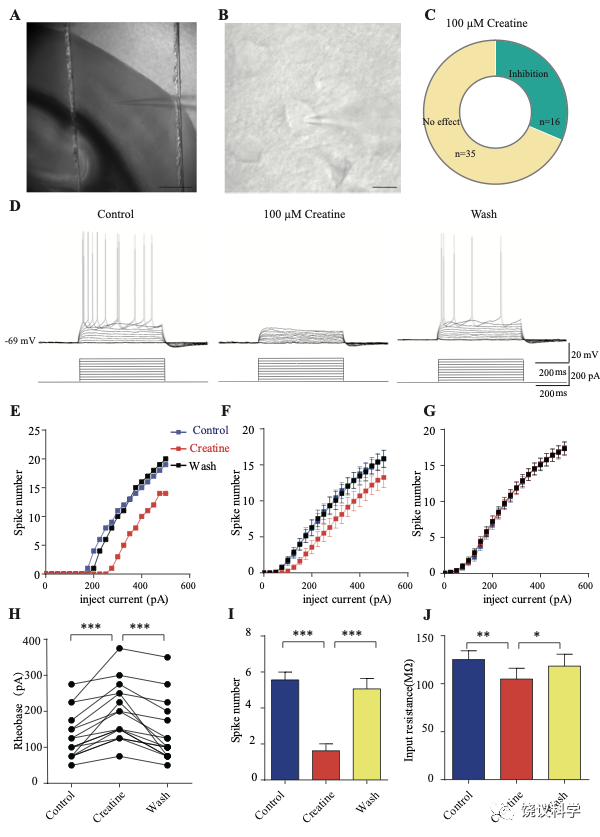
图7. 肌酐对皮层神经元的抑制作用. (A) A photograph showing recording at layer 4 in the somatosensory cortex. Scale bar: 10 μm. (B) Patch-clamp recording of a pyramidal neuron. Scale bar: 10 μm. (C) Ratios of Cr -responsive and -unresponsive neurons in the region. (D) Representative raw electrophysiological traces showing inhibition of evoked firing by Cr, with the lower panel showing the stimulus protocol. (E) Evoked spike numbers in response to different current injections from (D). (F) Relationship between evoked spike numbers and different current injections to neurons that were inhibited by Cr (n=16). (G) The same for Cr-unresponsive neurons (n=35). (H) Rheobase for Cr responsive neurons. (I) Evoked spike number when these neurons were injected with current of rheobase + 50 pA. (J) input resistance. *, p<0.05; **, p<0.01; ***, p<0.001, paired t test.
记录了膜电容(Cm) 114.96 ± 3.92 pF (n=51,图7—附图A)、中等大小椎体细胞(Figure 7B)。这些神经元在去极化的电流注射后发放有规律的电冲动201,202 (图7D和图7—附图3A),温和的最大诱发发放频率为每500毫秒(ms) 10-30次发放(图7 E-G),在去极化步骤之间发放间隔增加(图7D和图7—附图3A),高动作电位幅度 (81.64 ± 1.06 mV,图7—附图2E),以及大的发放半宽 (1.12 ± 0.031 ms,图7—附图2F)。
只在诱发发放模式达到稳态后,才在孵液中加肌酐。记录到的51个神经元中,16个被100 μM肌酐所抑制(图7 B-F)。在用肌酐期间,注射去极化电流(25 pA step, 500 ms)能够引起的对肌酐有反应的神经元发放冲动减少(图7 D-F)。肌酐的抑制作用可逆 (图7 D-F),通常在加入肌酐后2-3 min观察到作用 (最大作用在2到8 mins),而洗脱10到25 min后消失。第二次使用肌酐可以重复。阈强度(引起动作电位的最低电流),在给肌酐期间增加(图7H)。抑制作用最明显是在发放阈附近。用阈强度之上50 pA 电流去极化神经元时,如果用肌酐,诱导发放大大减少(图7I)。肌酐也能微弱地抑制输入电阻(图7J),稍微超极化静息膜电位 (图7—附图2C),降低第一次诱导动作电位后超极化(AHP)的幅度 (图7—附图2G)。发放阈值(图7—附图2D),幅度(图7—附图2E)和半宽(图7—附图2F) 等不为肌酐所影响。
其余35个神经元对肌酐没有反应(图7C和G,图7—附图3)。肌酐不改变其检测的电参数,包括发放频率(图7G,图7—附图3A, B和D),阈强度(图7—附图2C),静息膜电位(图7—附图2C),发放阈幅度(图7—附图2E) 和半宽 (图7—附图2F)。另外,反应的神经元与不反应的神经元在电特征上没有显著差别。以记录有些的神经元数量来看,第4曾和4/5层交界出反应神经元的比例似乎高于第5层深部(图7—附图1)。
突触小体摄取肌酐依赖于SLC6A8
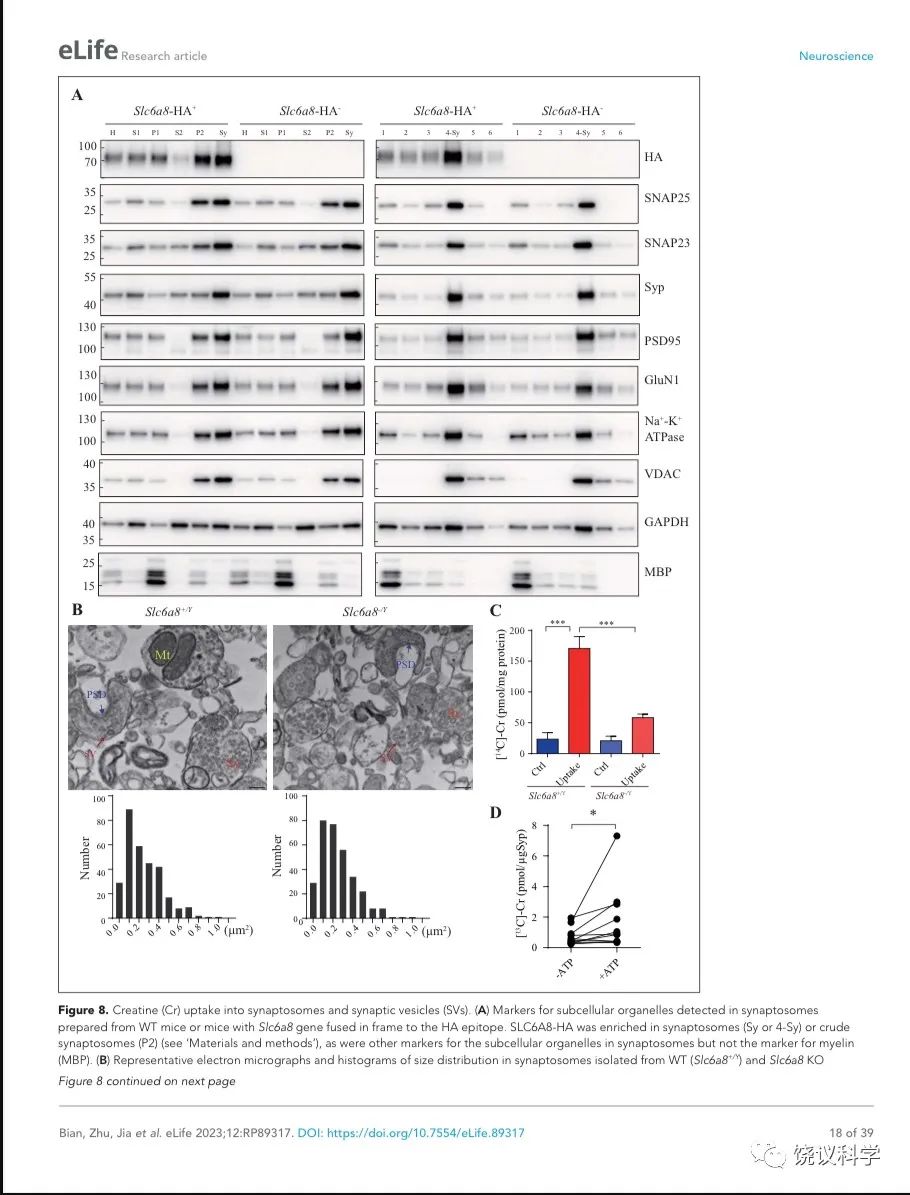
神经递质释放到突触间隙后,清除它们的方式有酶促降解和转运蛋白介导的再摄取。突触小体含神经传递的机构,常被用于研究神经递质再摄取203。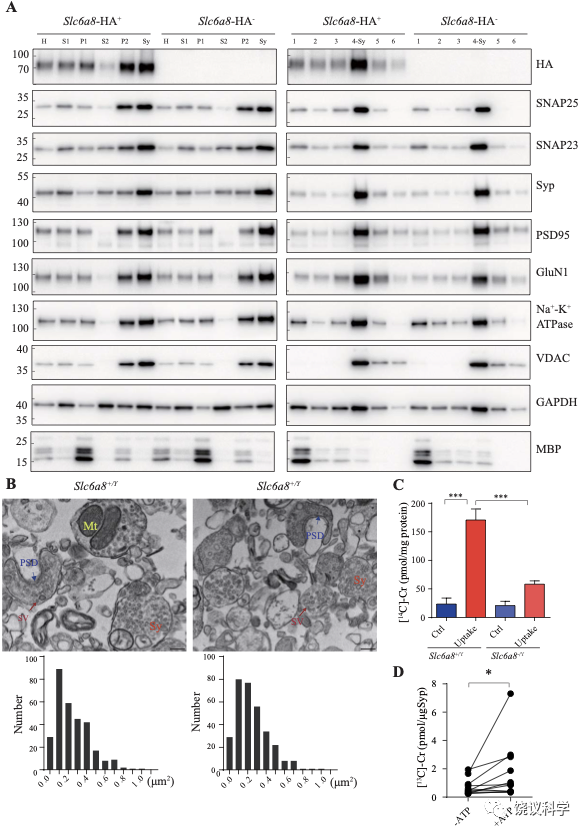
图8. 突触小体和SV摄取肌酐. (A) Markers for subcellular organelles detected in synaptosomes prepared from WT mice or mice with Slc6a8 gene fused in frame to the HA epitope. SLC6A8-HA was enriched in synaptosomes (Sy or 4-Sy) or crude synaptosomes (P2) (see methods), as were other markers for the subcellular organelles in synaptosomes but not the marker for myelin (MBP). (B) Representative electron micrographs and histograms of size distribution in synaptosomes isolated from WT (Slc6a8+/Y) and Slc6a8 KO (Slc6a8-/Y) mice by Ficoll density-gradient centrifugation. Sy, synaptosome; SV, synaptic vesicles; Mt, mitochondria; PSD, post-synaptic density. Bar, 20 nm. (C) Cr uptake into synaptosomes (n=5 per group). The two left columns were results from WT mice and the two right columns from Slc6a8 knockout mice. The control baseline was [14C]-Cr uptake at 0 ºC at 10 mins. Cr uptake into synaptosomes at 37 ºC measured at 10 mins was observed in WT synaptosomes. Uptake into Slc6a8 knockout synaptosomes was significantly reduced when compared to the WT synaptosomes. ***, p<0.001, one-way ANOVA with Tukey’s correction. (D) uptake of [13C]-Cr into immunoisolated SVs in the presence or absence of ATP. (n=11 samples per group)*, p<0.05, paired-t test.
为研究突触小体摄取肌酐是否依赖SLC6A8,我们先检测SLC6A8是否存在于突触小体中。用Slc6a8HA基因敲入小鼠和抗HA抗体,我们发现SLC6A8-HA存在于、并富集于突触小体粗提组分(图8A的P2组分,富集分:P2/H = 1.76 ± 0.15, n = 4),也存在于并富集于非连续Ficoll梯度获得的突触小体组分(图8A的Sy和4-Sy 等组分,富集分:Sy/H = 2.02 ±0.14, n = 4)。突触小体的完整性有多个突触小体标记物所确定203,包括突触前末梢的突触前细胞膜标记物SNAP25 204和SV标记Syp146-149,突触后致密标记PSD95 165,166和突触后膜蛋白GluN1 205,突触膜蛋白162-164, 细胞膜蛋白Na+-K+-ATPase 206-208,线粒体标记VDAC 203。这些都在突触小体制备中所富集。细胞浆标记GAPDH也存在与突触小体中,而少突胶质细胞标记MBP基本没有,提示很大程度被避免了髓磷脂污染203。
我们还用电镜验证了我们的突触小体制备质量。如前报道203,209,突触小体的组成为内充满SV的膜裹结构(图8B的Sy),有时有一段突触后膜包括突触后致密 (图8B的PSD)和线粒体 (图8B的Mt)。The sizes of synaptosomes from WT和Slc6a8基因剔除小鼠的突触小体大小相似,面积分别为0.245 ± 0.01 μm2 (n=302颗粒)和 0.247 ± 0.01 μm2 (n=317颗粒) (图8B)。
然后我们检测SLC6A8是否参与突触小体摄取肌酐。18 μM [14C]-肌酐 (总放射量为0.4 μCi)与5 μM肌酐混合,0 ºC、10 mins为基线210。与0 ºC (Ctrl,图8C) 相比,WT小鼠突触小体在37 ºC摄取肌酐约被刺激7倍(Uptake,图8C)。Cr uptake into synaptosomes from Slc6a8 基因剔除小鼠制备的突触小体摄取肌酐不到对照的3倍,只有WT小鼠的约三分之一(图8C)。所以,SLC6A8对于突触小体摄取肌酐是必需的。
SV摄取肌酐
经典神经递质被SV摄取依赖ATP144,159,181,210,211。我们检测了肌酐能否被SV所摄取。
我们用了10 μg的抗Syp抗体来纯化小鼠脑的SV。纯化的SV经过30 mins的预孵育以便内源肌酐充分漏出,然后与1 mM [13C]-肌酐混合,在有或五4 mM ATP的情况下,放在25 ℃ 孵育10 mins以便合适的摄取。用CE-MS和高效液相色谱(HPLC-MS)分析SV内含 [13C]-肌酐量。ATP存在时SV摄取[13C]-肌酐量显著提高,约10.3 pmol [13C]-肌酐转运进入SV(1.03 pmol/μg 抗Syp抗体,转运速度为0.103 pmol/min,n=11,图8D)。
总而言之,肌酐以ATP依赖的模式被转运进入SV。目前,我们不知道SV上的肌酐转运蛋白。SLC6A8仅见于细胞膜,未见于SV,不是SV摄取肌酐的转运蛋白候选分子。
讨论
虽然没有任何一个神经递质是由一篇论文所证明,本文所提高的支持肌酐作为可能新神经递质的证据达到了此前任何单一论文的程度。
在不同时候、由不同研究者怀疑过多个分子可能是神经递质,如牛磺酸 46,212、 脯氨酸 73、 D-天门冬氨酸213、 硫化氢 214、胍丁胺215、 多巴 216、 雌二醇 217、 beta-丙氨酸 218、质子219,但没有一个符合所有标准。有些被怀疑的分子可以被刺激诱导释放、或被转运蛋白移除。但常常他们不能可重复地在SV中检测到74。
我们在SV中发现肌酐显著地提高了检查肌酐作为候选分子的优先性。我们进一步的研究得到更多证据提示肌酐是神经递质: 1) Cr 储存于 SVs;2) 观察到了刺激引起的Ca++依赖的肌酐释放;3) SV储存肌酐和肌酐释放都在Slc6a8 或Agat 基因缺陷时降低;4) 肌酐抑制新皮层椎体神经元的活性; 5) 突触小体摄取肌酐需要SLC6A8;6) SV摄取肌酐依赖ATP。
以上结果中,1、3、4和6 在本文第一次发表。而且,我们显示了SV内含肌酐低于Glu和GABA,但高于ACh和5-HT,使肌酐水平置于已知中枢神经递质中等水平(图1-3)。SV存储肌酐依赖H+梯度(图1—附图2),肌酐可以被摄取如SV (图8D)。
以前有单独一篇文章报道电刺激后有Ca++依赖的[3H]肌酐释放、以及内源肌酐释放220。我们现在提供证据细胞外K+刺激(1-2 min之内)肌酐释放(图5-6)。而且肌酐释放会因为Slc6a8或Agat 基因敲除所降低(图5和6)。虽然K+诱导的Ca2+依赖的肌酐释低于Glu和GABA,但它存在而且可以因为Slc6a8 基因敲除而完全消失(图5)。以前报道的电诱导的肌酐释放显示更多Ca2+依赖性220。我们的资料与以前报道220支持肌酐为神经递质。我们观察到非常低的5-HT或ACh流出速率可能是因为切片中胆碱能神经元174 或五羟色胺能神经元176 数量有限,酶快速降解这些神经递质。
以前两次报道从细胞外空间摄取肌酐到细胞内,一次用脑片显示钠依赖的[3H]肌酐 220,一次用突触小体摄取肌酐221。我们新的肌酐不仅重复了突触小体肌酐摄取实验,而且显示它依赖表达在突触小体的膜转运蛋白SLC6A8(图8A)。SLC6A8转运肌酐入突触小体可能的功能包括从突触间隙清除肌酐、也可能将肌酐在循环到神经元的SV(图8B,图6D)。
总之,除了验证和延伸十年前孤独没有后续重复和追踪的结果,我们有全新的结果提示肌酐为神经递质。我们在下面讨论神经递质的标准,肌酐为神经递质以及肌酐作为神经递质的意义。
神经递质的标准
非肽类小分子作为神经递质的标准,因时间和作者不同而有所不同。
有些教科书简单地陈述:神经递质储存于突触前,刺激时释放,在突触后神经元显示活性。这三个标准的细节又不同。例如,有一本教科书要求“物质必须存在于突触前神经元;物质必须响应突触前去极化而释放,释放必须依赖于Ca2+; 突触后细胞必须有对于物质的特异受体”67,68。另一本教科书说:“分子必须在突触前神经元合成与储存;分子必须在刺激时有突触前轴突末端释放;实验给予分子必须在突触后细胞产生能够模拟突触前神经元刺激是否神经递质样的反应” 69。
过去四十年国际通用最广的神经科学教科书列了神经递质四个标准70,71:它在突触前神经元合成;它储存于囊泡,并释放足够的量作用与突触后神经元或靶器官;当加入合理浓度的外源分子,它可以模仿内源递质的作用;有特异机制从突触间隙清楚该物质。这些标准类似于、虽然不完全相同于神经递质的经典教科书所谓:神经递质“应该在突触前合成与释放;应该模拟神经刺激释放的内源分子的作用;如果可能,应该有药理激动剂或阻断剂,对内源释放分子的增强或阻断作用类似于对怀疑的神经递质的作用”222。另一教科书也列了药理标准223。
有作者注意到确立中枢神经递质的困难。例如,神经递质的专业书道“候选神经递质应该存在于突触前末梢,当突触前末梢活动时被释放,在实验施用时引起突触后神经元忠实的反应。实践中,因为中枢神经系统的神经元连续整合多种激动和抑制,最后一个标准被放宽到只要有活动的变化就可以”224。
经典神经递质、神经肽及其受体的领袖科学家,写道:“定一个分子为递质取决于所用标准,最常见的是物质在神经元合成,从其末梢释放,模拟生理神经传递的效果,具有灭活的机制。但是,每一个新的候选分子的出现导致规则的修改和变宽”225。
肌酐作为神经递质的证据
经典教科书列了16个小分子神经递质70,71。其中的腺苷、花生四烯酸、一氧化氮、二氧化碳目前不符合所有四个标准。肌酐看上去比这些更符合中枢神经递质的标准。
我们这篇文章得到的结果显示肌酐满足了Robinson 224的中枢神经递质标准。
Snyder及其同事225 的四个标准大都符合,只是上了神经传递还需要更多研究,因为需要确定特异的突触并研究可能的肌酐能神经传递。这在中枢神经系统要比外周神经系统需要长很多的时间。有些通常接受为神经递质的从未在严格意义上满足这一条件。肌酐移除机制的标准不仅因脑片肌酐摄取220和突触小体肌酐摄取221所满足,而且因为我们显示突触小体摄取肌酐需要SLC6A8。
Kandel 等70,71 的四个标准大都被满足,但有细节需要进一步研究。合成的标准通常不是很严格,因为有些递质是在某些细胞合成后转运到其他细胞起递质作用。我们发现肌酐存在于SV中就可以替代合成的要求,因为存在于神经元的SV提供了足够证据,肌酐位于正确的地点可以起到神经递质的作用。SV肌酐含量高于ACh和5-HT (图1和2)。肌酐释放量与Glu和GABA在同一数量级(图5和6)。特异移除机制的标准由脑片肌酐摄取实验220 和突触小体实验221所满足,进一步由我们发现SLC6A8参与突触小体肌酐摄取的结果所加强(图8)。
在此我们报道在与经典神经递质可比的浓度抑制小鼠特定脑区的椎体细胞,大约1/3椎体细胞对100 mM肌酐有反应(图7)。以前的报道中,100 μM到2 mM 的GABA226-228、50 μM到2 mM的Glu 229,230、1到100 μM的DA229、0.1到100 μM的5-HT231-236以孵育投放来研究神经递质的生理功能。我们的结果揭示,给孵育液加入100 μM肌酐后几分钟内可以抑制皮层神经元,时程类似于5-HT237 234 236和DA 229,但显著慢于Glu230、GABA48,227和5-HT233。
最近一篇报道,基因敲除Slc6a8提高皮层神经元的兴奋性267。前额叶皮层(PFC)椎体细胞的电生理分析发现诱发放电频率增加。因为我们显示肌酐可以抑制新皮层部分椎体神经元(Figure 8),那篇文章提供的体内证据与肌酐可能是抑制性神经元的可能性一致。
肌酐与经典神经递质的差别
目前,我们没有肌酐的分子确定受体,仅从肌酐引起的电生理反应推测其存在。我们猜想肌酐可能作用于G蛋白偶联受体(GPCRs),而不是快速作用的配体门控的离子通道,如Glu的AMPA或NMDA受体和GABA的GABAA受体。以前有过关于肌酐对神经元作用的报道,包括称肌酐是GABAA受体部分激动剂238-241。 这些作用需要很高浓度的肌酐 (十毫摩尔量级)。也有一篇相反作用的报道:肌酐 (浓度超过500 μM)在60 mins后通过NMDA受体增加神经元的兴奋性,时程显著慢于经典神经递质242。
细胞外K+诱导的非Ca2+依赖性肌酐释放较Glu和GABA明显的多。一个可能性是非Ca2+依赖性肌酐释放来自于神经胶质细胞,因为GAMT被报道高度表达于星状胶质细胞261和少突细胞261, 262。如以前报道,其他神经调质如牛磺酸可以由星状胶质细胞263或脑片264以非Ca2+依赖性释放。另外,在没有钾刺激时, Ca2+耗竭可以增加体外培养的星状胶质细胞265或纹状体266释放牛磺酸。类似地, Slc6a8 KO脑片中,Ca2+耗竭(图5G)也增加肌酐的基线,相比于正常ACSF培养液(图5D)。
因为有长的多的研究历史,现在在其他方面ACh和5-HT有多于肌酐的证据是中枢递质,特别是ACh和5-HT有很多激动剂、拮抗剂证明有些教科书222,223需要、有些教科书不需要的更多的一条神经递质标准。药理标准需要更多时间和努力,因为,迄今为止,没有人试图找过肌酐的激动剂或拮抗剂。
对SLC6A8和肌酐意味着什么
值得注意SLC6A8属于神经递质转运蛋白家族SLC,已经有多个成员被证明转运神经递质65,66,90-99。
其他人和我们做的摄取实验显示SLC6A8在脑内将肌酐转运入神经元。AGAT也表达与脑内,但不是表达SLC6A8的细胞 243,244。肌酐及其前体被认为在神经系统内不同细胞之间转运。当完全缺失SLC6A8时,如Slc6a8缺陷纯合子病人,肌酐治疗没有效果。而SLC6A8部分有功能时,肌酐有效245。Slc6a8突变杂合体女性的难治性癫痫被肌酐完全治好246。我们发现肌酐对皮层神经元的抑制作用提供了解释其抗癫痫作用的新机制247。
因为星状胶质细胞的伪足围绕微毛细血管内皮细胞(MCEC)形成血脑屏障(BBB),星状胶质细胞不表达Slc6a8提示脑内肌酐不依赖外周转运进入,而主要在脑内合成80,82,248。SLC6A8在脑内转运肌酐及其前体,而不是跨BBB转运肌酐的主要蛋白。按我们用突触小体的实验结果(图8) 221,它转运肌酐入突触前末梢。
已知肌酐有能量来源的其他效果,补充肌酐被认为有助于儿童、孕妇和哺乳妇女、以及老人79,249。肌酐被报道可以改善人的脑力活动250-255。肌酐被用于潜在治疗动物的神经退行性变模型256,257。
我们的工作会刺激进一步研究,区分以前怀疑的肌酐效果哪些不是因其能量储存的作用,而是其神经递质作用。
寻找新的神经递质
我们的工作可刺激寻找更多的神经递质。我们的发现表明几十年前停了寻找神经递质是技术原因,不是因为缺乏更多神经递质。大多数已知小分子神经递质是因为其外周效果而发现的事实也支持缺乏的是有组织的努力来寻找没有外周效果的中枢神经递质。新的神经递质可能从长期怀疑的候选分子钟发现,也可以从以前没有怀疑的分子中发现,甚至可能从以前未知的分子中发现。
需要创新的途径揭示没有以前怀疑和线索的分子。高度纯化的SV,不同脑区的SV,含特异SLC的SV提供未来研究的有些起点。
致谢
我们感谢章晓辉、张新祥、罗敏敏、郭青春、刘以群、胡迎春、和戈鹉平博士的讨论、建议和技术帮助, Jiang Chen, Jing Cai, Yulin Jiang, Jinhuan Ou, Xiaomeng Deng, Jiawen Liu和Meng Wu的技术支持, 北京脑研究所、北大清华生命科学联合中心、昌平实验室、中国医科院 (2019RU003)的支持。饶实验室的研究从未被中国脑计划所污染。
伦理
所有动物实验都得到北京大学动物中心的同意。实验按照北京大学动物使用委员会(IACUC)的规范和批准(批准号LSC-RaoY-9 and LSC-RaoY-10 )。
附图及图例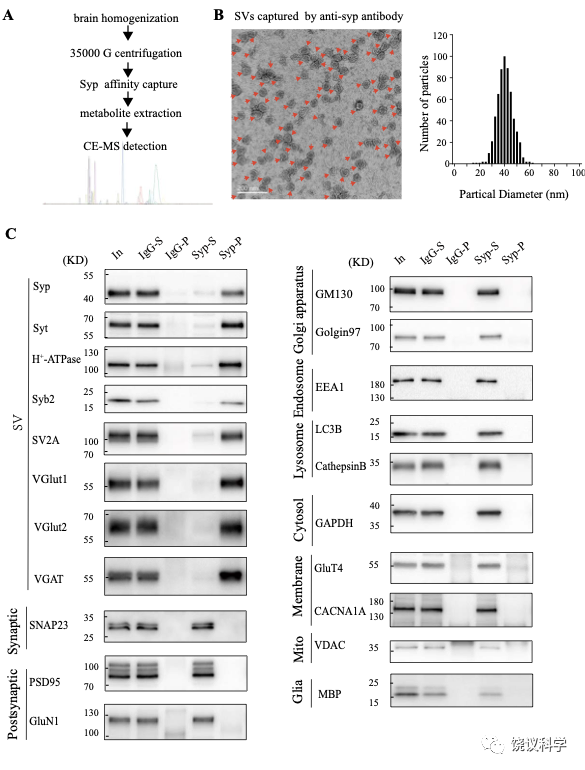
图1—附图1. 验证小鼠脑SV的纯化. (A) A diagram for SV purification and SV content analysis. Brain homogenates were centrifugated at 35000 g for 25 mins and supernatants containing SVs were collected as the starting material. Following immunopurification by the anti-Syn antibody, SVs were broken by water to release their contents. Metabolites were analyzed by CE-MS. (B) EM of SVs captured by the anti-Syn antibody. Arrows pointing to SVs, the size each being 40.4 ± 0.26 nm, with size distribution shown on the right. (C) Analysis of SV purity by immunoblotting. Markers for SVs (Syp, syt, H+-ATPase, syb2, SV2A, VGLU1, VGLU2 and VGAT) were effectively pulled down by beads coated with the anti-Syn antibody but not by those coated with IgG. Non-SV markers present in supernatants, including those for the lysosome (LAMP1, cathepasinB, LC3B), the Golgi apparatus (GM130, Golgi 97), the early endosome (EEA1), the cytoplasma (GAPDH), synaptic membrane (SNAP23) and postsynaptic components (PSD95, GluN1), the mitochondria (VDAC), the cytoplasmic membrane (CACNA1A), axonal membrane (GluT4), and glia membrane (MBP), could not be pulled down by either the anti-Syp antibody or IgG. 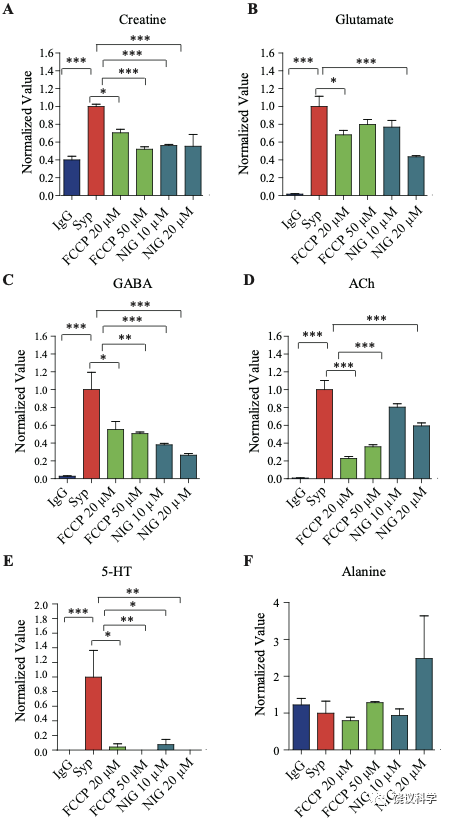
图1—附图2. 药理抑制剂对SV内含的效果. (A) Cr (B) Glu (C) GABA (D) ACh (E) 5-HT (F) Alanine*, p<0.5, **p<0.05, ***, p<0.01, one-way ANOVA with Tukey’s correction (n=4 per group). Data were normalized to average amount of molecules pulled down by the anti-Syp antibody. NIG, nigrecin.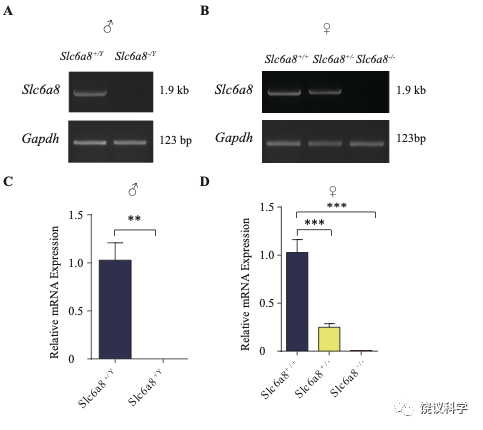
图2—附图2. Slc6a8基因敲除小鼠的体重和脑重. (A) Brain weights of male mice. (B) Body weights of male mice. (C) Brain weight of female mice. (D) Body weights of female mice. **, p<0.01 in (B), student t test. *, p<0.05, **, p<0.01,*** in (e), p<0.001, one-way ANOVA with Tukey’s correction (n=5 mice per group, 7 weeks old).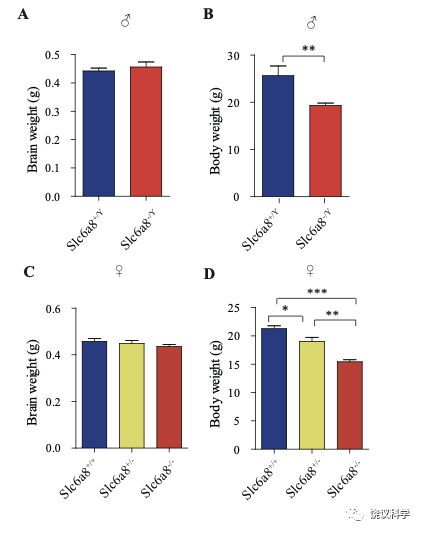
图2—附图2. Slc6a8基因敲除小鼠的体重和脑重. (A) Brain weights of male mice. (B) Body weights of male mice. (C) Brain weight of female mice. (D) Body weights of female mice. **, p<0.01 in (B), student t test. *, p<0.05, **, p<0.01,*** in (e), p<0.001, one-way ANOVA with Tukey’s correction (n=5 mice per group, 7 weeks old).
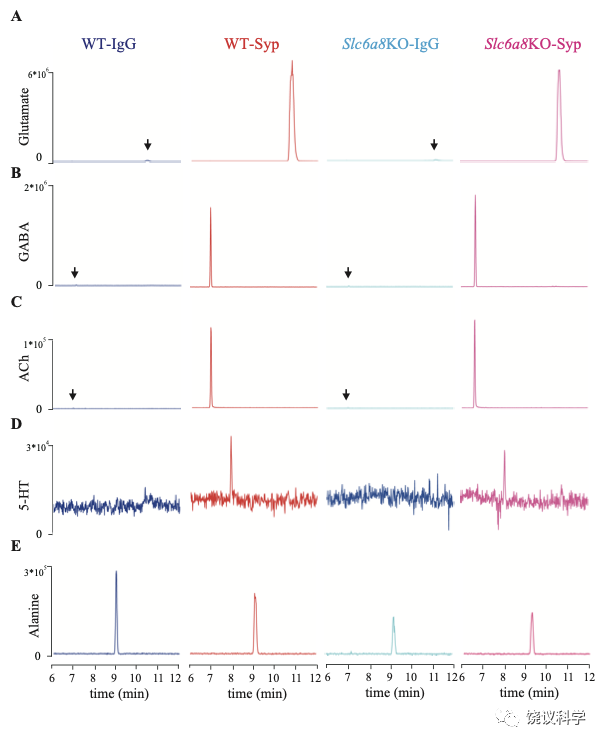
图3. 分子的代表性CE-MS资料. (A-E) Molecules pull-down from WT mice by IgG (WT-IgG), anti-Syp antibody (WT-Syp), and those from knockout mice (Slc6a8 KO-IgG, Slc6a8 KO-Syp). (A) Glu. (B) GABA. (C) ACh. (D) 5-HT. (E) Alanine. Vertical: signal amplitude; abscissa: retention time.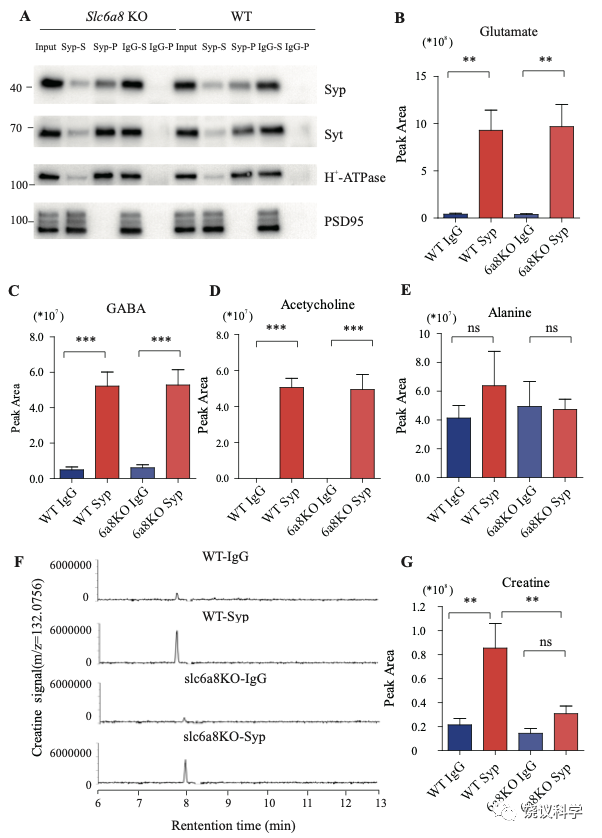
图2—附图4. 野生型和Slc6a8基因敲除型小鼠SV中检测到的蛋白质和小分子. (A) Immunoblotting analysis of SV markers Syp, syt, H-ATPase and postsynaptic components (PSD95). (B-D) Peak areas of CE-MS signals for neurotransmitters: Glu, GABA, and ACh. These neurotransmitters are enriched in SVs and not affected by Slc6a8genotypes. (E) Alanine was not enriched in SVs of either WT or Slc6a8 KO mice. (F) Representative raw example traces of Cr. (G) Cr in SVs was lower in Slc6a8 KO mice than that in WT mice. **, p<0.01, ***, p<0.001, one-way ANOVA with Tukey’s correction (n=8 samples per group). 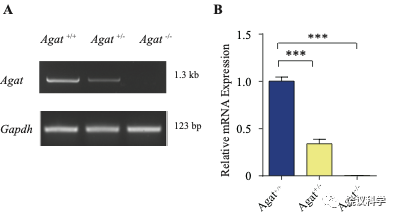
图3—附图1. 验证Agat-KO first 小鼠. (A) RT-PCR analysis of the entire coding region showing the absence of Agat mRNA in Agat-/- mice and decreased expression in Agat+/- mice. (B) Quantitative RT-PCR analysis. ***, p<0.001, one-way ANOVA with Tukey’s correction (n=4 mice per group, with 3 repeats for each mice). 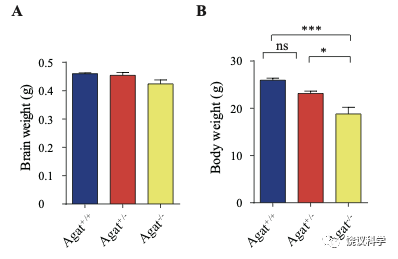
图3—附图2. Agat+/+, Agat+/- 和Agat-/- 小鼠的体重和脑重. (A) Brain weights of male mice. (B) Body weights of male mice. *, p<0.05, **, p<0.01, * in (E), p<0.001, one-way ANOVA with Tukey’s correction, ns, not significant. (n=5 mice per group, 7- weeks).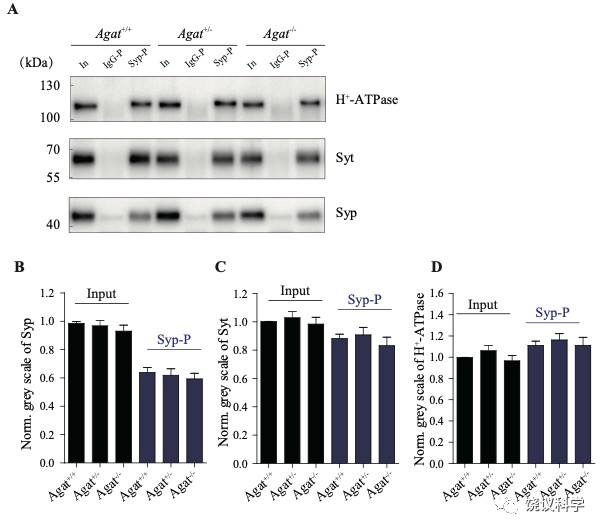
图3—附图3. Agat+/+, Agat+/- 和Agat-/- 小鼠含类似量的SV. (A) Representative Western analysis of SV markers H+-ATPase, Syt and Syp from input, samples pulled down by IgG (IgG-P) and anti-Syp antibody (Syp-P). (B-E) Quantification of protein band densities for Syp (B), Syt (C) and H+-ATPase (D) (n=20 per group). 
图7—附图1. 记录位点在体躯感觉皮层的分布. (A) High levels of SLC6A8-HA signal observed in the somatosensory cortex, especially in layer 4. (B) Recording sites. Red denotes the responsive neurons and blue the unresponsive neurons. 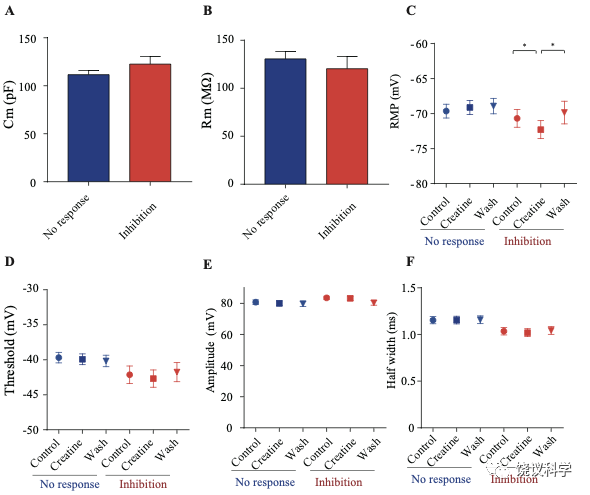
图7—附图2.肌酐反应(蓝)和肌酐不反应 (红)神经元的电生理参数. There was no significant difference between the two groups of neurons in membrane capacitance (Cm) (A) and membrane resistance (Rm) (B), resting membrane potential (RMP) (C), spike threshold (D), spike amplitude (E), half width of spike (F) and amplitude of after hyperpolarization (AHP) (G). While no electrical parameters detected were changed by Cr in non-responsive neurons (n=35), resting membrane potential (C) was slightly inhibited by Cr and AHP (G) was slightly elevated (n= 16) in Cr responsive neurons (*, p<0.05, paired t test).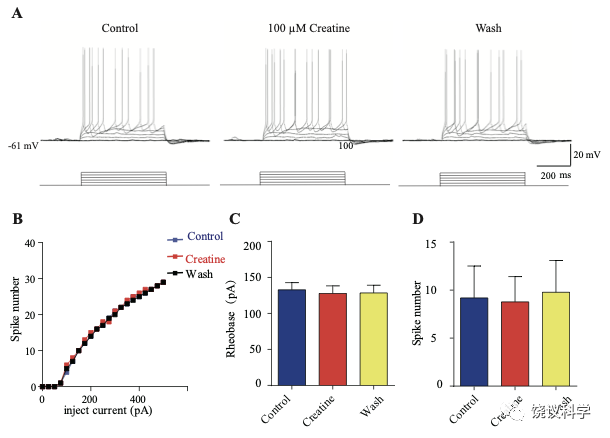
图7—附图3. 肌酐不反应神经元的电生理资料. (A) Representative raw traces. (B) I-V curve of this same neuron. No effect of 100 μM Cr on rheobase current and evoked spike number at a current of 50 pA above rheobase current was observed. (C-D) Rheobase and spike number were not changed during Cr application.
材料与方法
制造基因敲除和基因敲入小鼠.
Slc6a8 knockout and knockin mice were generated using CRISPR-Cas9-mediated genome engineering techniques by Beijing Biocytogen (Beijing, China). Agat ‘knockout-first’ 185 mice were purchased from CAM-SU GRC (Suzhou, China). All mutations were validated by southern blot analysis, tail junction PCR and DNA sequencing. Transgenic mice will be provided upon request.
反向转录多聚酶反应(RT-PCR)和定量实时PCR(qPCR).
Total RNA of whole brains from mice of different genotypes was extracted using the Buffer RZ (Tiangen, no. RK14, Beijing, China) and reverse transcribed into complementary DNA (cDNA) using the RevertAid First-Strand cDNA synthesis kit (Thermo Scientific, K1622, Waltham, Massachusetts,USA). qPCR was performed using the Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Q712-02) on Bio-Rad CFX-96 Touch Real-time PCR System (Bio-Rad, USA). Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as an internal control. ΔCt (difference in cycle threshold) was calculated for each sample (ΔCt = Ct Target gene − Ct GAPDH) for further evaluation of relative mRNA expression levels in different genotypes. The sequence specificities of the primers were examined. Three pairs of primers targeting different genes were used: Slc6a8 forward, 5’-GTCTGGTGACGAGAAGAAGGG-3’, Slc6a8 reverse, 5’- CCACGCACGACATGATGAAGT-3’; Agat forward, 5’-CACAGTGGAGGTGAAGGCCAATACATAT-3’, Agatreverse, 5’-CCGCCTCACGGTCACTCCT-3’; Gapdh forward, 5’- AGGTCGGTGTGAACGGATTTG-3’, Gapdhreverse, 5’- TGTAGACCATGTAGTTGAGGTCA-3’.
Primers for reverse PCR were designed to obtain complete coding sequences based on information obtained from National Center for Biotechnology Information (NCBI): Slc6a8 forward, 5’-ATGGCGAAAAAGAGCGCTGAAAACG-3’; Slc6a8 reverse, 5’-TTACATGACACTCTCCACCACGACGACC-3’; Agat forward, 5’- ATGCTACGGGTGCGGTGTCT-3’; Agat reverse, 5’- TCAGTCAAAGTAGGACTGAAGGGTGCCT-3’. PCR products were electrophoresed on 1% agarose gels, stained with GelRed, visualized under UV illumination and photographed.
免疫印迹分析.
Samples were loaded onto 10% polyacrylamide gels with the PAGE system (#1610183, Bio-Rad Laboratories, USA) and run in the SDS running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.8) for 25 min at 80 V followed by 25 - 45 min at 200 V. Afterward, proteins were transferred to immobilon NC transfer membranes (HATF00010, Millipore) at 400 mA for 2 h in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol). Membranes were blocked in 5% fat-free milk powder in TBST (25 mM Tris, 150 mM NaCl, 0.2%Tween-20 (P1397, Sigma, St. Louis, Missouri, USA), pH 7.4 adjusted with HCl) and incubated overnight with the indicated primary antibodies dissolved in TBST containing 2% BSA.
Primary antibodies are listed below: rabbit anti-Synaptophysin (dilution 1:5000, cat no. 101002, synaptic systems(SySy), Goettingen,Germany), rabbit anti-Synaptotagmin1/2 (dilution 1:2000, cat no.105002, SySy, Germany), rabbit anti-proton ATPase (dilution 1:1000, cat no.109002, SySy, Germany), rabbit anti-Synaptobrevin 2 (dilution 1:5000, cat no.104202, SySy, Germany), rabbit anti-SV2A (dilution 1:2000, cat no.109003, SySy, Germany), rabbit anti-VGlut1(dilution 1:4000, 135302, SySy, Germany), rabbit anti-VGlut2 (dilution 1:2000, 135402, SySy, Germany), rabbit anti-VGAT(dilution 1:4000, 131002, SySy, Germany), rabbit anti-SNAP23 (dilution 1:2000, cat no. 111202, SySy, Germany), mouse anti-PSD95 (dilution 1:5000, cat no 75028, NeuroMab, Davis, Calornia, USA ), mouse anti-GluN1(dilution 1:5000, cat no 114011, SySy, Germany), rabbit anti-GM130 (dilution 1:1000, cat no ab52649, Abcam), rabbit anti-Golgin-97 (dilution 1:2000, cat no13192, Cell Signaling Technology, Danvers, Massachusetts, USA), rabbit anti-EEA1 (dilution 1:2000, cat no 3288, Cell Signaling Technology, Danvers, Massachusetts, USA), rabbit anti-LC3B (dilution 1:1000, cat no 2775S , Cell Signaling Technology, Danvers, Massachusetts, USA), goat anti-CathepsinB (dilution 1:2000, AF965, R&D Systems, Minneapolis, Minnesota, USA), rabbit anti-GAPDH (dilution 1:1000, cat no 2118S, Cell Signaling Technology, Danvers, Massachusetts, USA), rabbit anti-GluT4 (dilution 1:1000, ab33780, Abcam, Cambridge, UK), rabbit anti-CACNA1A (dilution 1:300, 152103, SySy, Germany), rabbit anti-VDAC (dilution 1:1000, cat no 4661S , Cell Signaling Technology, Danvers, Massachusetts, USA), rabbit anti-MBP (1:1000 , cat no. 295003, SySy, Germany), mouse anti Creatine Kinase B (dilution 1:5000, cat no MAB9076, , Minneapolis, Minnesota, R&D Systems), rabbit anti HA (dilution 1:2000, CST3724; Cell Signaling Technology, Danvers, Massachusetts, USA) and rabbit anti SNAP25 (dilution 1:2000, ab109105, Abcam, UK) antibodies.
Membranes were washed in three washing steps in TBST (each for 5 mins) and incubated with peroxidase-conjugated secondary antibodies for 2-3 h at 4 ºC. The second antibody used were anti-rabbit (dilution 1:5000, A6154, Sigma, St. Louis, Missouri, USA), anti-mouse (dilution 1:5000, 715-035-150, Jackson Immuno Research, West Grove, Philadelphia, USA) or rabbit anti-goat IgG secondary antibodies (dilution 1:1000, cat no ab6741, Abcam, UK). After repeated washing, signals were visualized using a ChemiDoc XRS+ System (Bio-RAD Laboratories, USA).
分离突触囊泡.
Our purification procedures for synaptic vesicles were based on previously established immunoisolation methods 143,151. Protein G magnetic beads (cat no.88848, Thermofisher Scienctific, Waltham, Massachusetts, USA) were washed three times with IP buffer (100 mM potassium tartrate, 4 mM HEPES-KOH, 2 mM MgCl2, pH 7.4) supplemented with a complete protease inhibitor cocktail (Roche, Basel, Switzerland). 5 μg monoclonal anti-Syp antibody directed against a cytoplasmic epitope (cat no 101011, SySy, Germany) or control mouse IgG (10400C, Thermofisher Scientific, Waltham, Massachusetts, USA) was used to incubate with 20-30 μl beads for 30 mins at RT in 2% BSA dissolved in IP buffer. Under this condition, 4-4.5 μg of antibody was coupled, as determined by Western blot and Commassie Blue staining. Immunoisolation of SVs were carried out at 0 °C-2 °C to prevent vesicular content leakage (with RT as a control). Briefly, the whole mouse brain was homogenized in 3 ml of IP buffer with a glass/Teflon homogenizer (20 strokes at 2000 rpm, WHEATON, USA and WIGGENS WB2000-M, Germany) immediately after decapitation. Homogenates were centrifuged for 25 mins at 35,000 x g and the supernatant was adjusted to approximately 3 mg/ml protein (Nanodrop 2000 C, Thermofisher Scientific,Waltham, Massachusetts, USA). To capture the SVs for content detection, about 200 μl of supernatants (per 5 μg anti-Syp/IgG) were incubated with pre-coupled beads for 2.25 h under slow rotation at 2 °C. Beads were washed 6 times for further Western blot analysis and vesicular content detection. For pharmacological blockade of H+-gradient across SV membrane, the mix of supernatants and pre-coupled beads was diluted into 1.2 ml before the addition of inhibitors.
确定囊泡内涵.
To extract SV contents, immunoisolates were treated with 50 μl ultra-pure water. 100 μl methanol together with 100 μl acetonitrile were added to precipitate proteins in samples. After centrifugation for 20 mins at 168000 × g, supernatants were collected and centrifuged for 20 mins at 2000 × g to remove beads and proteins. Samples were pre-frozen with liquid nitrogen and vacuum dried at -45 °C overnight. Dried samples were kept frozen and re-suspended with 50 μl of 0.2 μM 13C-creatine (internal control) immediately before detection.
Capillary electrophoresis-mass spectrometry (CE-MS) was used to verify and quantify small molecules. CE/MS detection was applied with the coupling of PA800 plus CE system (Beckman Coulter, Brea, CA, USA) and mass spectrometry (TRIPLE QUAD 5500, AB SCIEX or Q Exactive HF-X, Thermo Scientific, Waltham, Massachusetts, USA). Before SV content detection, we optimized MS detection of classical neuro transmitters, Cr and amino acids in positive ion mode. Firstly, the fragment ions (Q3) for a given molecule (precursor ions, Q1) were determined by either systematic scanning of standard sample solution (0.1 μM in 10% acetate acid) or referring to database (www.mzcloud.org). Secondly, optimal values of collision energy (CE), collision cell exit potential (CXP) and declustering potentials (DP) were determined for each pair of Q1/Q3. Thirdly, optimal combination of parameters (Q1/Q3, CE, CXP, DP) was chosen for each molecule. In addition, parameters were adjusted every 2 to 3 months for best signal to noise ratios.
CE/MS separations were carried out by capillaries (OptiMS silica surface cartridge, Beckman Coulter, Brea, CA, USA). The CE background electrolyte was 10 % acetate acid. Each new separation capillary was activated with rinsing under 100 psi sequentially with methanol for 10 mins (forward), methanol for 3 mins (reverse), H2O for 10 mins (forward), H2O for 3 mins (reverse), 0.1 M NaOH for 10 mins (forward), water for 5 mins (reverse), 0.1 M HCl for 10 mins (forward), followed by water for 10 mins and then 10 % acetate acid for 10 mins (forward) and 3 mins (reverse), prior to the first use. Between analyses, the capillary was rinsed with 10% acetate acid under a 100 psi pressure for 5 min (forward) flowed by 75 psi for 4 min. The sample (50 μl) was injected with 2.5-4 psi for 30 s. Separation voltage of 25 kV was applied for 25 mins. To maintain stably spray during CE separation, ion spray voltage was applied at 1.7-1.9 kV. MS data were collected 5 mins after CE separation. Finally, the capillary was washed with 10% acetate acid for 10 mins, followed by methanol for 20 min and then10% acetate acid for 20 mins.
Standard solutions of 0.2 μM 13C-Cr (internal control) and analytes were used to plot standard curves. Linear standard curves (R2>0.98, for most cases, R2>0.99), calculated from peak area ratios corresponding to analytes and internal standards, were obtained for all molecules tested. The concentration ranges used for standards of Glu, GABA, ACh, 5-HT, Cr and alanine were 0.03-10 μM, 0.003-1 μM, 0.0003-0.1 μM, 0.003-1 μM, 0.03-1 μM and 0.03-1 μM, respectively. Standard curves were made at least twice for a given capillary. Analytes of SV contents were calculated using the standard curves and normalized to the amount of anti-Syp antibody conjugated to the beads.
电子显微镜.
All EM grids were glow discharged for 30 s using a plasma cleaner (Harrick PDC-32G-2, plasma cleaners, Ithaca, New York). To free SVs from beads, 25 μl 0.1 M glycine-HCl (PH=2) was incubated for 1 min and quickly neutralized with 25 μl 0.1 M Tris (pH=10). Beads were quickly removed and 2-4 μL aliquots of SVs were applied to the carbon coated copper grids (Zhong Jing Ke Yi, Beijing,China). After 1 min, the grid was dried with a filter paper (Whatman No.1), and placed in the water, and then immediately stained using 2 % uranyl acetate for 30 s. At last uranyl acetate was removed and the grid was air dried. The grids were examined on a JEM-F200 electron microscope (JEOL,Tokyo, Japan) operated at 200 kV. Images were recorded using a 4k × 4k COMS One view camera (Gatan, Abingdon, UK). Fixation of synaptosomal pellets was performed by immersion with pre-warmed 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at RT for 2 h. After washing 4 times with 0.1 M phosphate buffer (pH 7.4) every 15 mins, samples were post-fixed with 1% osmium tetroxide (w/v) at 4°C for 1 h and then washed 3 times. Following en bloc staining with 2% uranyl acetate (w/v) at 4°C overnight, samples were dehydrated and embedded in fresh resin, polymerized at 65°C for 24 h. Ultrathin (70 nm) sections were obtained by Leica UC7 ultramicrotome (Leica Microsystems, wetzlar, German) and recorded on 80 kV in a JEOL Jem-1400 transmission electron-microscope (JEOL,Tokyo,Japan) using a CMOS camera (XAROSA, EMSIS, Munster, Germany).
免疫组织化学.
Adult mice were anesthetized by i.p. injection with 2 % 2,2,2-tribromoethanol (T48402, Sigma, St. Louis, Missouri, USA) in saline at a dose of 400 mg/kg, and perfused trancardially with 0.9 % saline followed by 4 % PFA in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH=7.4).
Brains were cryoprotected with 30 % sucrose in30 % sucrose 0.1 M PB (81 mM Na2HPO4, 19 mM NaH2PO4) and sectioned in the coronal plane (40 μm thick) using a Cryostat (Leica 3050S, wetzlar, German). For anti-HA immunostaining, we used a rabbit monoclonal anti-HA antibody (1:500 in 0.3 % Triton in PBS; 48 h incubation at 4 °C; #3724, Cell Signaling Technology, Danvers, Massachusetts, USA), followed by a goat anti-rabbit Alexa Fluor 546 secondary antibody (1:1000; overnight at 4 °C; # A-11035, Invitrogen, Waltham, Massachusetts, USA). Sections were mounted in a medium containing 50 % glycerol, cover-slipped, and sealed with nail polish. Images were acquired using virtual slide microscope (Olympus VS120-S6-W, Tokyo, Japan) and a laser-scanning confocal microscope (Zeiss 710, Cambridge, UK) and brain structures inferred with an established mouse brain atlas 258.
制备脑片.
Male C57 mice (of 30-38 days-old) were anesthetized with pentobarbital (250 mg/kg) and decapitated. Brains were quickly removed and placed into ice-cold, low calcium, high magnesium artificial cerebrospinal fluid (ACSF) with sodium replaced by choline. The medium consisted of 120 mM choline chloride, 2.5 mM KCl, 7 mM MgSO4, 0.5 mM CaCl2, 1.25 mM NaH2PO4, 5 mM sodium ascorbate, 3 mM sodium pyruvate, 26 mM NaHCO3, and 25 mM D-(+)-glucose, and was pre-equilibrated with 95% O2-5% CO2. Coronal brain slices (300 μm thick) were cut with a vibratome (Leica VT1200S, wetzlar, German). Slices were incubated for 1 h at 34°C with oxygenated ACSF containing: 124 mM NaCl, 2.5 mM KCl, 2 mM MgSO4, 2.5 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM D- (+)-glucose.
诱发脑片释放.
Coronal brain slices (each 300 μm thick, typically with a wet weight of 17-20 mg) were transferred into a specially designed superfusion chamber with a volume of approximately 200 μl, containing freshly 95% O2/5% CO2 oxygenated ACSF. Slices were equilibrated for 10 min in ACSF at a superfusion rate of 0.9-1.25 ml/min. The “control” sample was collected for 1 min just before high K+ stimulation (K-ACSF, 70 mM KCl replacing equal amount of NaCl). We waited for 30 seconds to allow K+ stimulus to immerse the slices (dead volume for solution transition of 200 μl and chamber volume of 200 μl), then the sample “70 mM K” in response to K-ACSF was collected for another 1 min. Following 10 min of washout period, we collected the third sample of “wash” for 1 min.
To detect Ca2+ dependent release, slices were pre-incubated for 10 mins with normal ACSF and equilibrated with Ca2+ free ACSF (containing 1 mM EGTA to chelate extracellular Ca2+) for 10 mins. The baseline sample “0 Ca2+ ACSF” was collected for 1 min. Superfusion solution was changed to Ca2+ free K-ACSF for 2 mins and sample “0 Ca2+ 70 mM K” was collected (dead volume for solution transition of 400 μl and chamber volume of 200 μl). After that, the solution was changed back to normal ACSF for 10 mins and K-ACSF for 2 mins. The sample “2.5 mM Ca 70 mM K” for the last min was collected.
Samples were subjected to CE-MS in a method similar to SV content detection, except for the following: 1) standards were dissolved in ACSF or other buffers used in release experiment; 2) concentration ranges used for standards of Glu was from 0.003-1 μM. 3) To protect the MS from salt pollution, data were collected from 10 mins - 20 mins during CE separation.
膜片钳记录.
Slices were transferred to a recording chamber on an upright fluorescent microscope equipped with differential interference contrast optics (DIC; Olympus BX51WI). Slices were submerged and superfused with ACSF at about 2.8 ml/min at 24-26 °C. Whole cell patch recordings were routinely achieved from layer 4/5 medium sized pyramidal neurons from the somatosensory cortex. Patch pipettes (3-5 MΩ) contained: 140 mM K-gluconate, 10 mM HEPES, 0.5 mM EGTA, 5 mM KCl, 3 mM Na2-ATP, 0.5 mM Na3GTP, and 4 mM MgCl2 (with pH adjusted to 7.3 and osmolarity of 290 mOsm/kg). Current-clamp recordings were carried out with a computer-controlled amplifier (Multiclamp 700B, Molecular Devices) and traces were digitized at 10 kHz (DigiData 1550B, Molecular Devices). Data were collected and analyzed using Clampfitor Clampex 10 software (Molecular Devices).
Cells were characterized by their membrane responses and firing patterns during hyperpolarizing and depolarizing current steps (-100 to +500 pA, increment: 50 pA or 25 pA, 500 ms). Regular spiking pyramidal neurons were identified by moderate maximal spiking frequencies (20-60 Hz, that is, 10-30 spikes per 500 ms, Figure 7E-G), increasing of inter-spike intervals during depolarizing step (Figure 7B andFigure 7—figure supplement 3A), high action amplitude (Figure 7—figure supplement 2E) and large half width (Figure 7—figure supplement 2F) 201,202. After the mean firing frequency evoked by current injections reached the steady state for at least 5 mins (typically 20-30 mins following the formation of whole-cell configuration), 100 μM Cr was bath-applied for 6 mins. Typically, Cr was applied for a 2nd time following washout to re-confirm the effects.
突触小体制备.
Synaptosomes were isolated by Ficoll/sucrose density-gradient centrifugation 203,209,221,259. Whole brains from adult male mice were homogenized with 15 strokes at 900 rpm in buffer A (320 mM sucrose, 1 mM EDTA, 1mM EGTA, 10 mM Tris–HCl, pH 7.4, with a complete protease inhibitor cocktail (Roche, Basel, Switzerland)). The homogenate (H fraction) was centrifuged at 1000 g for 10 min to precipitate the membrane fragments and nuclei (P1 fraction). Supernatant was centrifuged again at 1000 g for 10 min and the resulting supernatant (S1) was centrifuged at 12,000 g for 20 min. Supernatant was the S2 fraction, and the pellet was re-suspended with buffer A and centrifuged at 12,000 g for 20 min. The resulting pellet was crude synaptosomes (P2 fraction), containing synaptosomes with mitochondria and microsomes.
Crude synaptosomes (P2 fraction) was re-suspended with 150-200 μl buffer B (320 mM sucrose and 10 mM Tris–HCI (pH 7.4)). The sample was carefully overlaid on the top of a gradient of 2 ml of 7.5 % (wt/vol in buffer B) Ficoll and 1.8 ml of 13% (wt/vol in buffer B) Ficoll and centrifuged at 98,000 g for 45 min at 2-4 °C in a swinging-bucket rotor. A myelin band was present near the surface, and th synaptosomes band (fraction Sy) was present at the interface between the 13% and 7.5 % Ficoll layers, with the mitochondria being pelleted at the bottom. For further western analysis, the supernatant was divided into 6 fractions (600 μl for each fraction) and the mitochondria pellet was discarded. The isolated synaptosomes was included in fraction 4.
For Western analysis, fractions H, S1, P1, S2, P2 and Sy were adjusted to 0.5 mg/ml by bicinchoninic acid assay (BCA) method with reference to NanoDrop™ 2000 Spectrophotometers. 3.35 μg protein was loaded for each lane. Fractions 1- 6 were loaded with the same volume (10 μl composed of 6.7 μl sample and 3.3 μl loading buffer) for each lane.
突触小体肌酐摄取.
To remove Ficoll, we diluted the synaptosomal band (480 μl) with 4.3 ml of a pH 7.4 buffer C containing (in mM) 240 mannitol, 10 glucose, 4.8 potassium gluconate, 2.2 Calcium gluconate, 1.2 MgSO4, 1.2 KH2PO4 and 25 HEPES-Tris. The sample was then centrifuged at 12,000 g and the pellet was re-suspended with buffer C. Uptake experiments were either performed at 37ºC or at 0ºC (control). For each sample, 25-43 μg of synaptosomes (with a volume of 40-50 μl) were added to 360 μl buffer containing (in mmol/L) 100 NaCl, 40 mannitol, 10 glucose, 4.8 potassium gluconate, 2.2 Calcium gluconate, 1.2 MgSO4, 1.2 KH2PO4, 25 HEPES and 25 Tris (pH adjusted to 7.4). A mixture of 18 μM [14C]-creatine (0.4 μCi) and 5 μM creatine was quickly added. After 10 min, uptake was terminated by the addition of 1 ml of NaCl-free ice-cold buffer C. Samples were immediately filtered, under vacuum, through a Whatman GF/C glass filter (1825-025) pre-wetted with buffer C. Filters were further washed with 10 ml of ice-cold buffer C, dissolved in scintillation fluid and the radioactivity determined by liquid scintillation spectrometry.
SV摄取肌酐.
The uptake of 13C-creatine was assayed according to a conventional procedure 260 with slight modifications: the immunoisolated synaptic vesicles by 10 μg Syp antibody (101011, SySy, Germany) were resuspended with the uptake buffer (150mM meglumine-tartrate, 4 mM KCl, 4 mM MgSO4, 10 mM HEPES-KOH (pH 7.4 ), and complete EDTA-free protease inhibitor cocktail) containing 4 mM Mg-ATP or additional 4 mM MgSO4, followed by preincubation for 30 min at 25 ℃. The uptake reaction was started by addition of 1 mM 13C-creatine dissolved in the uptake buffer with a final volume of 125 μL (pH at 6.8). After 10 min at 25℃, 1 mL of ice-cold uptake buffer was added to the incubation to stop the reaction, followed by 5 more times washing. The SV contents were extracted using the protocol described in the determination of vesicular contents part. 100 nM Cr was used as the internal control. CE-MS and LC-MS was used to verify and quantify the creatine contents of samples. A Vanquish UHPLC system coupled to a Q Exactive HF-X mass spectrometer (both instrument from Thermo Fisher Scientific, Waltham, Massachusetts, USA) were used for LC-MS analysis along with SeQuant ZIC-HILIC column (150 mm × 2.1 mm, 3.5 μm, Merck Millipore, 150442) in the positive mode and SeQuant ZIC-pHILIC column (150 mm × 2.1 mm, 5 μm, Merck Millipore, 150460) in the negative mode. For ZIC-HILIC column, the mobile phase A was 0.1% formic acid in water and the mobile phase B was 0.1% formic acid in acetonitrile. The linear gradient was as following: 0 min, 80% B; 6 min, 50% B; 13 min, 50% B; 14 min, 20% B; 18 min, 20% B; 18.5 min, 80% B; 30 min, 80% B. The flow rate used was 300 μl/min and the column temperature was maintained at 30°C. For ZIC-pHILIC column, the mobile phase A is 20 mM ammonium carbonate in water, adjusted to pH 9.0 with 0.1% ammonium hydroxide solution (25%) and the mobile phase B is 100% acetonitrile. The linear gradient was as follows: 0 min, 80% B; 2 min, 80% B; 19 min, 20% B; 20 min, 80% B; 30 min, 80% B. The flow rate used is 150 μl/min and the column temperature is 25°C. Samples were maintained at 4°C in Vanquish autosampler. 3 µl of extracted metabolites were injected for each run. IP samples were subjected to ZIC-HILIC column in positive mode for major metabolites detection, and then subject to ZIC-pHILIC column in negative mode for orthogonal detection.
参考文献
1. Du Bois-Reymond, E. H. Gesammelte Abhandlungen zur Allgemeinen Muskel-und Nervenphysik. (Veit & Co, 1877).
2. Langley, J. N. On the stimulation and paralysis of nerve-cells and of nerve-endings: Part I. J Physiol 27, 224-236 (1901).
3. Elliot, T. R. On the action of adrenalin. J Physiol 31, xx-xxi (1904).
4. Elliot, T. R. The action of adrenalin. J Physiol 32, 401-467, doi:10.1113/jphysiol.1905.sp001093 (1905).
5. Dale, H. H. The action of certain esters and ethers of choline, and their relation to muscarine. Journal of Pharmacology and Experimental Therapeutics 6, 147-190 (1914).
6. Loewi, O. Über humorale Übertragbarkeit der Herznervenwirkung. I. Pflüger Archiv Physiol 189, 239-242 (1921).
7. Rao, Y. The First Hormone: Adrenaline. Trends Endocrinol Metab 30, 331-334 (2019).
8. Claude, B. Leçons sur les effets des substances toxiques et médicamenteuses. (JB Baillière, 1857).
9. Langley, J. N. & Dickinson, W. L. On the Local Paralysis of Peripheral Ganglia, and on the Connexion of Different Classes of Nerve Fibres with Them. Proc R Soc Lond 46, 423-431 (1889).
10. Langley, J. N. On the reaction of cells and of nerve-endings to certain poisons, chiefly as regards the reaction of striated muscle to nicotine and to curari. J Physiol 33, 374-413 (1905).
11. Langley, J. N. On nerve endings and on special excitable substances in cells. Proceedings of the Royal Society of London Series B-Containing Papers of a Biological Character 78, 170-194 (1906).
12. Strecker, A. Ueber einige neue Bestandtheile der Schweinegalle. Justus Liebigs Annalen der Chemie 123, 964-965 (1862).
13. Liebreich, O. Ueber die chemische Beschaffenheit der Gehirnsubstanz. Justus Liebigs Annalen der Chemie und Pharmacie 134, 29-44 (1865).
14. Baeyer, A. Mittheilungen aus dem organischen Laboratorium de Gewerbeacademie zu Berlin. I. Über das Neurin (Notes from the organic laboratory of the Gewerbeacademie in Berlin. I on the neurin). . Justus Liebig’s Annalen der Chemie 142, 322-326 (1867).
15. Hunt, R. & Taveau, R. D. M. On the physiological action of certain cholin derivatives and new methods for detecting cholin. Br Med J 1906, 1788-1789 (1906).
16. Mott, F. W. & Halliburton, W. D. The Physiological Action of Choline and Neurine. Br Med J 1, 1082-1083 (1899).
17. Hunt, R. Note on a blood pressure lowering body in the suprarenal gland. Am J Physiol 3, xviii-xix (1900).
18. Ewins, A. J. Acetylcholine, a New Active Principle of Ergot. Biochem J 8, 44-49 (1914).
19. Dale, H. H. & Dudley, H. W. The presence of histamine and acetylcholine in the spleen of the ox and the horse. J Physiol 68, 97-123 (1929).
20. Feldberg, W. & Gaddum, J. H. The chemical transmitter at synapses in a sympathetic ganglion. J Physiol 81, 305-319 (1934).
21. Brown, G. L. & Feldberg, W. The acetyloholine metabolism of a sympathetic ganglion. J Physiol 88, 265-283 (1936).
22. Dale, H. H., Feldberg, W. & Vogt, M. Release of acetylcholine at voluntary motor nerve endings. J Physiol 86, 353-380 (1936).
23. Addison, T. On the constitutional and local effects of disease of the supra-renal capsules. (Highley, 1855).
24. Schäfer, E. A. ON THE PRESENT CONDITION OF OUR KNOWLEDGE REGARDING THE FUNCTIONS OF THE SUPRARENAL CAPSULES. Br Med J 1, 1277-1281 (1908).
25. Oliver, G. & Schäfer, E. A. The Physiological Effects of Extracts of the Suprarenal Capsules. J Physiol 18, 230-276 (1895).
26. Abel, J. J. On epinephrin, the active constituent of the suprarenal capsule and its compounds. Proc Am Physiol Soc3, 4-5 (1898).
27. Abel, J. J. & Crawford, A. C. On the blood pressure raising constituent of the suprarenal capsule. Johns Hopkins Hosp Bull 8, 151-157 (1897).
28. Abel, J. J. Über den blutdruckerregenden Bestandtheil der Nebenniere, das Epinephrin. Hoppe-Seyler’s Zeitschrift für physiolgische Chemie 28, 318-362 (1899).
29. Abel, J. J. Further observations on epinephrine. Johns Hopkins Hosp Bull 12, 80-84 (1901).
30. Takamine, J. Adrenalin, the active principle of the suprarenal glands and its mode of preparation. Am J Pharmacy73, 523-531 (1901).
31. Takamine, J. The isolation of the active principle of the suprarenal gland. J Physiol 27, 29-30 (1902).
32. Aldrich, T. B. A preliminary report on the active principle of the suprarenal gland. Am J Physiol 5, 457-461 ( 1901).
33. Langley, J. N. Observations on the physiological action of extracts of the supra-renal bodies. J Physiol 27, 237-256 (1901).
34. Barger, G. & Dale, H. H. Chemical structure and sympathomimetic action of amines. J Physiol 41, 19-59 (1910).
35. Cannon, W. B. & Rosenblueth, A. Sympathin E and Sympathin I. Am J Physiol 104, 557-574 (1933).
36. Euler, U. S. v. A specific sympathomimetic ergone in adrenergic nerve fibers (sympathin) and its relation to adrenaline and noradrenaline. Acta Physiol Scand 12, 73-97 (1946).
37. Euler, U. S. v. Identification of the sympathomimetic ergone in adrenergic nerves of cattle (Sympathin N) with laevo-noradrenaline. Acta Physiol Scand 16, 63-74 (1948).
38. Euler, U. S. v. Noradrenaline. Chemistry, physiology, pharmacology and clinical aspects. (Springfield, 1956).
39. Mitchell, J. F. The spontaneous and evoked release of acetylcholine from the cerebral cortex. J Physiol 165, 98-116 (1963).
40. Collier, B. & Mitchell, J. F. The central release of acetylcholine during stimulation of the visual pathway. J Physiol184, 239-254 (1966).
41. Collier, B. & Mitchell, J. F. The central release of acetylcholine during consciousness and after brain lesions. J Physiol 188, 83-98 (1967).
42. Carlsson, A., Falck, B. & Hillarp, N. A. Cellular localization of brain monoamines. Acta Physiol Scand 56(Suppl), 1-28 (1962).
43. Björklund, A., Ehinger, B. & Falck, B. A method for differentiating dopamine from noradrenaline in tissue sections by microspectrofluorometry. J Histochem Cytochem 16, 263-270 (1968).
44. Robbins, J. The effects of amino acids on the crustacean neuro-muscular system. Anat Rec 132, 492-493 (1958).
45. Curtis, D. R., Phillis, J. W. & Watkins, J. C. Chemical excitation of spinal neurones. Nature 183, 611-612 (1959).
46. Curtis, D. R. & Watkins, J. C. The excitation and depression of spinal neurones by structurally related amino acids. J Neurochem 6, 117-141 (1960).
47. Curtis, D. R. & Watkins, J. C. Acidic amino acids with strong excitatory actions on mammalian neurones. J Physiol 166, 1-14 (1963).
48. Roberts, E. & Frankel, S. γ-Aminobutyric acid in brain: its formation from glutamic acid. J Biol Chem 187, 55-63 (1950).
49. Udenfriend, S. Identification of gamma-aminobutyric acid in brain by the isotope derivative method. J Biol Chem187, 65-69 (1950).
50. Florey, E. An inhibitory and an excitatory factor of mammalian central nervous system, and their action of a single sensory neuron. Archieves Internationales de Physiologie 62, 33-53 (1954).
51. Florey, E. & McLennan, H. The release of an inhibitory substance from mammalian brain, and its effect on peripheral synaptic transmission. J Physiol 129, 384-392 (1955).
52. Fatt, P. & Katz, B. Some observations on biological noise. Nature 166, 597-598(1950).
53. Fatt, P. & Katz, B. Spontaneous subthreshold activity at motor nerve endings. J Physiol 117, 109-128 (1952).
54. Del Castillo, J. & Katz, B. Quantal components of the end-plate potential. J Physiol 124, 560-573 (1954).
55. Robertson, J. D. Ultrastructure of two invertebrate synapses. Proc Soc Exp Biol Med 82, 219-223 (1953).
56. Palade, G. E. & Palay, S. L. Electron microscope observations of interneuronal and neuromuscular synapses. Anat Rec 118, 335-336 (1954).
57. Palay, S. L. Electron microscope study of the cytoplasm of neurons. Anat Rec 118, 336 (1954).
58. De Robertis, E. D. P. & Bennett, H. S. Sub-microscopic vesicle component in the synapse. Fed Proc 13, 35 (1954).
59. De Robertis, E. D. P. & Bennett, H. S. Some features of the sub-microscopic morphology of the synapses in frog and earthworm. J Biophys Biochem Cytol 1, 47–55 (1955).
60. Del Castillo, J. & Katz, B. Biophysical aspects of neuro-muscular transmission. Prog Biophys 6, 121-170 (1956).
61. Axelrod, J., Weil-Malherbe, H. & Tomchick, R. The physiological disposition of H3-epinephrine and its metabolite metanephrine. J Pharmacol Exp Ther 127, 251-256 (1959).
62. Radian, R. & Kanner, B. I. Reconstitution and purification of the sodium- and chloride-coupled gamma-aminobutyric acid transporter from rat brain. J Biol Chem 260, 11859-11865 (1985).
63. Radian, R., Bendahan, A. & Kanner, B. I. Purification and identification of the functional sodium- and chloride-coupled gamma-aminobutyric acid transport glycoprotein from rat brain. J Biol Chem 261, 15437-15441 (1986).
64. Guastella, J. et al. Cloning and expression of a rat brain GABA transporter. Science 249, 1303-1306 (1990).
65. Pacholczyk, T., Blakely, R. D. & Amara, S. G. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature 350, 350-354 (1991).
66. Blakely, R. D. et al. Cloning and expression of a functional serotonin transporter from rat brain. Nature 354, 66-70 (1991).
67. Purves, D., Augustine, G.J., Firzpatrick, D., Katz, L., LaMantia, A.-S., McNamara, J.O.,, and Williamss, S.M. Neuroscience, 2nd edition. (Sinauer Associates, 2001).
68. Purves, D., Augustine, G.J., Fitzpatrick, D., Hall, W.C., LaManita, A.-S., Mooney, R.D., Platt, M.L., and White, L.E. . Neuroscience, 6th edition. (Sinauer Associates, 2016).
69. Bear, M. F., Connors, B. W. & Paradiso, M. A. Neuroscience, Exploring the Brain. 4th Edition. (Wolters Kluwer, 2016).
70. Kandel, E. R., Schwartz, J.M., Jessell, T.M., Siegelbaum, S.A., and Hudspeth, A.J. Principles of Neural Sciences. 5th Edition. (McGraw-Hill, 2013).
71. Kandel, E. R., Koester, J.D., Mack, S.H., and Siegelbaum, S.A. Principles of Neural Sciences. 6th Edition., (McGraw-Hill, 2021).
72. Siegel, A. & Sapru, H. N. Essential Neuroscience, 2nd edition. (Wolters Kluwer, 2011).
73. Felix, D. & Künzle, H. Iontophoretic and autoradiographic studies on the role of proline in nervous transmission. Pflugers Arch 350, 135-144 (1974).
74. Chantranupong, L. et al. Rapid purification and metabolomic profiling of synaptic vesicles from mammalian brain. Elife 9, e59699 (2020).
75. Chevreul, E. Sur la composition chimique du bouillon de viandes. J Pharm Sci Access 21, 231-242 (1835).
76. Liebig, J. Kreatin und Kreatinin, Bestandtheile des Harns der Menschen. Journal für Praktische Chemie 40, 288-292 (1847).
77. Wyss, M. & Kaddurah-Daouk, R. Creatine and creatinine metabolism. Physiol Rev 80, 1107-1213 (2000).
78. Brosnan, J. T. & Brosnan, M. E. Creatine: endogenous metabolite, dietary, and therapeutic supplement. Annu Rev Nutr 27, 241-261 (2007).
79. Wallimann, T., Tokarska-Schlattner, M. & Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 40, 1271-1296 (2011).
80. Braissant, O., Henry, H., Béard, E. & Uldry, J. Creatine deficiency syndromes and the importance of creatine synthesis in the brain. Amino Acids 40, 1315-1324 (2011).
81. Ohtsuki, S. et al. The blood-brain barrier creatine transporter is a major pathway for supplying creatine to the brain. J Cereb Blood Flow Metab 22, 1327-1335 (2002).
82. Braissant, O., Henry, H., Loup, M., Eilers, B. & Bachmann, C. Endogenous synthesis and transport of creatine in the rat brain: an in situ hybridization study. Mol Brain Res 86, 193-201 (2001).
83. Braissant, O., Bachmann, C. & Henry, H. Expression and function of AGAT, GAMT and CT1 in the mammalian brain. Subcell Biochem 46, 67-81 (2007).
84. Nelson, N. The family of Na+/Cl- neurotransmitter transporters. J Neurochem 71, 1785-1803 (1998).
85. Chen, N. H., Reith, M. E. & Quick, M. W. Synaptic uptake and beyond: the sodium- and chloride-dependent neurotransmitter transporter family SLC6. Pflugers Arch 447, 519-531 (2004).
86. Höglund, P. J., Adzic, D., Scicluna, S. J., Lindblom, J. & Fredriksson, R. The repertoire of solute carriers of family 6: identification of new human and rodent genes. Biochem Biophys Res Commun 336, 175-189 (2005).
87. Bröer, S. & Gether, U. The solute carrier 6 family of transporters. Br J Pharmacol 167, 256-278 (2012).
88. Iversen, L. Neurotransmitter transporters and their impact on the development of psychopharmacology. Br J Pharmacol 147 Suppl 1, S82-88 (2006).
89. Bröer, S. The SLC6 orphans are forming a family of amino acid transporters. Neurochem Int 48, 559-567 (2006).
90. Guastella, J., Brecha, N., Weigmann, C., Lester, H. A. & Davidson, N. Cloning, expression, and localization of a rat brain high-affinity glycine transporter. Proc Natl Acad Sci U S A 89, 7189-7193 (1992).
91. Clark, J. A., Deutch, A. Y., Gallipoli, P. Z. & Amara, S. G. Functional expression and CNS distribution of a beta-alanine-sensitive neuronal GABA transporter. Neuron 9, 337-348 (1992).
92. Borden, L. A., Smith, K. E., Hartig, P. R., Branchek, T. A. & Weinshank, R. L. Molecular heterogeneity of the gamma-aminobutyric acid (GABA) transport system. Cloning of two novel high affinity GABA transporters from rat brain. J Biol Chem 267, 21098-21104 (1992).
93. Lopez-Corcuera, B., Liu, Q. R., Mandiyan, S., Nelson, H. & Nelson, N. Expression of a mouse brain cDNA encoding novel gamma-aminobutyric acid transporter. J Biol Chem 267, 17491-17493 (1992).
94. Giros, B., el Mestikawy, S., Bertrand, L. & Caron, M. G. Cloning and functional characterization of a cocaine-sensitive dopamine transporter. FEBS Lett 295, 149-154 (1991).
95. Kilty, J. E., Lorang, D. & Amara, S. G. Cloning and expression of a cocaine-sensitive rat dopamine transporter. Science 254, 578-579 (1991).
96. Shimada, S. et al. Cloning and expression of a cocaine-sensitive dopamine transporter complementary DNA. Science 254, 576-578 (1991).
97. Hoffman, B. J., Mezey, E. & Brownstein, M. J. Cloning of a serotonin transporter affected by antidepressants. Science 254, 579-580 (1991).
98. Smith, K. E., Borden, L. A., Hartig, P. R., Branchek, T. & Weinshank, R. L. Cloning and expression of a glycine transporter reveal colocalization with NMDA receptors. Neuron 8, 927-935 (1992).
99. Liu, Q. R., Nelson, H., Mandiyan, S., López-Corcuera, B. & Nelson, N. Cloning and expression of a glycine transporter from mouse brain. FEBS Lett 305, 110-114 (1992).
100. Mayser, W., Schloss, P. & Betz, H. Primary structure and functional expression of a choline transporter expressed in the rat nervous system. FEBS Lett 305, 31-36 (1992).
101. Guimbal, C. & Kilimann, M. W. A Na(+)-dependent creatine transporter in rabbit brain, muscle, heart, and kidney. cDNA cloning and functional expression. J Biol Chem 268, 8418-8421 (1993).
102. Gonzalez, A. M. & Uhl, G. R. 'Choline/orphan V8-2-1/creatine transporter' mRNA is expressed in nervous, renal and gastrointestinal systems. Mol Brain Res 23, 266-270 (1994).
103. Nash, S. R. et al. Cloning, pharmacological characterization, and genomic localization of the human creatine transporter. Rec Chan 2, 165-174 (1994).
104 . Schloss, P., Mayser, W. & Betz, H. The putative rat choline transporter CHOT1 transports creatine and is highly expressed in neural and muscle-rich tissues. Biochem Biophys Res Commun 198, 637-645 (1994).
105. Sora, I. et al. The cloning and expression of a human creatine transporter. Biochem Biophys Res Commun 204, 419-427 (1994).
106. Barnwell, L. F., Chaudhuri, G. & Townsel, J. G. Cloning and sequencing of a cDNA encoding a novel member of the human brain GABA/noradrenaline neurotransmitter transporter family. Gene 159, 287-288 (1995).
107. Happe, H. K. & Murrin, L. C. In situ hybridization analysis of CHOT1, a creatine transporter, in the rat central nervous system. J Comp Neurol 351, 94-103 (1995).
108. Saltarelli, M. D., Bauman, A. L., Moore, K. R., Bradley, C. C. & Blakely, R. D. Expression of the rat brain creatine transporter in situ and in transfected HeLa cells. Dev Neurosci 18, 524-534 (1996).
109. Mak, C. S. et al. Immunohistochemical localisation of the creatine transporter in the rat brain. Neuroscience 163, 571-585 (2009).
110. Speer, O. et al. Creatine transporters: a reappraisal. Mol Cell Biochem 256, 407-424 (2004).
111 . Lowe, M. T., Faull, R. L., Christie, D. L. & Waldvogel, H. J. Distribution of the creatine transporter throughout the human brain reveals a spectrum of creatine transporter immunoreactivity. J Comp Neurol 523, 699-725 (2015).
112. Salomons, G. S. et al. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am J Hum Genet 68, 1497-1500 (2001).
113. Mancardi, M. M. et al. Severe epilepsy in X-linked creatine transporter defect (CRTR-D). Epilepsia 48, 1211-1213 (2007).
114. Battini, R. et al. Language disorder with mild intellectual disability in a child affected by a novel mutation of SLC6A8 gene. Mol Genet Metab 102, 153-156 (2011).
115. Rosenberg, E. H. et al. High prevalence of SLC6A8 deficiency in X-linked mental retardation. Am J Hum Genet75, 97-105 (2004).
116. Newmeyer, A., Cecil, K. M., Schapiro, M., Clark, J. F. & Degrauw, T. J. Incidence of brain creatine transporter deficiency in males with developmental delay referred for brain magnetic resonance imaging. J Dev Behav Pediatr 26, 276-282 (2005).
117. Clark, A. J. et al. X-linked creatine transporter (SLC6A8) mutations in about 1% of males with mental retardation of unknown etiology. Hum Genet 119, 604-610 (2006).
118. Lion-François, L. et al. High frequency of creatine deficiency syndromes in patients with unexplained mental retardation. Neurology 67, 1713-1714 (2006).
119. Arias, A. et al. Creatine transporter deficiency: prevalence among patients with mental retardation and pitfalls in metabolite screening. Clin Biochem 40, 1328-1331 (2007).
120. Puusepp, H. et al. The screening of SLC6A8 deficiency among Estonian families with X-linked mental retardation. J Inherit Metab Dis 33, S5-11 (2010).
121. Cheillan, D. et al. Screening for primary creatine deficiencies in French patients with unexplained neurological symptoms. Orphanet J Rare Dis 7, 96 (2012).
122. van de Kamp, J. M., Mancini, G. M. & Salomons, G. S. X-linked creatine transporter deficiency: clinical aspects and pathophysiology. J Inherit Metab Dis 37, 715-733(2014).
123. Joncquel-Chevalier Curt, M. et al. Creatine biosynthesis and transport in health and disease. Biochimie 119, 146-165 (2015).
124. DesRoches, C. L. et al. Estimated carrier frequency of creatine transporter deficiency in females in the general population using functional characterization of novel missense variants in the SLC6A8 gene. Gene 565, 187-191 (2015).
125. Skelton, M. R. et al. Creatine transporter (CrT; Slc6a8) knockout mice as a model of human CrT deficiency. PLoS One 6, e16187 (2011).
126. Kurosawa, Y. et al. Cyclocreatine treatment improves cognition in mice with creatine transporter deficiency. J Clin Invest 122, 2837-2846 (2012).
127. Baroncelli, L. et al. A mouse model for creatine transporter deficiency reveals early onset cognitive impairment and neuropathology associated with brain aging. Hum Mol Genet 25, 4186-4200 (2016).
128. Udobi, K. C. et al. Cognitive deficits and increases in creatine precursors in a brain-specific knockout of the creatine transporter gene Slc6a8. Genes Brain Behav 17, e12461 (2018).
129. Molinaro, A. et al. A Nervous System-Specific Model of Creatine Transporter Deficiency Recapitulates the Cognitive Endophenotype of the Disease: a Longitudinal Study. Sci Rep 9, 62 (2019).
130. Abdulla, Z. I. et al. Deletion of the Creatine Transporter (Slc6a8) in Dopaminergic Neurons Leads to Hyperactivity in Mice. J Mol Neurosci 70, 102-111 (2020).
131 . Stöckler, S. et al. Creatine deficiency in the brain: a new, treatable inborn error of metabolism. Pediatr Res 36, 409-413 (1994).
132. Bianchi, M. C. et al. Reversible brain creatine deficiency in two sisters with normal blood creatine level. Ann Neurol 47, 511-513 (2000).
133. Item, C. B. et al. Arginine:glycine amidinotransferase deficiency: the third inborn error of creatine metabolism in humans. Am J Hum Genet 69, 1127-1133 (2001).
134. Stockler-Ipsiroglu, S. et al. Guanidinoacetate methyltransferase (GAMT) deficiency: outcomes in 48 individuals and recommendations for diagnosis, treatment and monitoring. Mol Genet Metab 111, 16-25 (2014).
135. Stockler-Ipsiroglu, S. et al. Arginine:glycine amidinotransferase (AGAT) deficiency: Clinical features and long term outcomes in 16 patients diagnosed worldwide. Mol Genet Metab 116, 252-259 (2015).
136. Khan, A., Tian, L., Zhang, C., Yuan, K. & Xu, S. Genetic diversity and natural selection footprints of the glycine amidinotransferase gene in various human populations. Sci Rep 6, 18755 (2016).
137. Fons, C. & Campistol, J. Creatine Defects and Central Nervous System. Semin Pediatr Neurol 23, 285-289 (2016).
138. Whittaker, V. P., Michaelson, I. A. & Kirkland, R. J. The separation of synaptic vesicles from nerve-ending particles ('synaptosomes'). Biochem J 90, 293-303 (1964).
139. Nagy, A., Baker, R. R., Morris, S. J. & Whittaker, V. P. The preparation and characterization of synaptic vesicles of high purity. Brain Res 109, 285-309 (1976).
140. Matthew, W. D., Tsavaler, L. & Reichardt, L. F. Identification of a synaptic vesicle-specific membrane protein with a wide distribution in neuronal and neurosecretory tissue. J Cell Biol 91, 257-269 (1981).
141. De Camilli, P., Harris, S. M., Jr., Huttner, W. B. & Greengard, P. Synapsin I (Protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J Cell Biol 96, 1355-1373 (1983).
142. Huttner, W. B., Schiebler, W., Greengard, P. & De Camilli, P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96, 1374-1388(1983).
143. Burger, P. M. et al. Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron 3, 715-720 (1989).
144. Burger, P. M. et al. GABA and glycine in synaptic vesicles: storage and transport characteristics. Neuron 7, 287-293 (1991).
145. Maycox, P. R., Link, E., Reetz, A., Morris, S. A. & Jahn, R. Clathrin-coated vesicles in nervous tissue are involved primarily in synaptic vesicle recycling. J Cell Biol 118, 1379-1388 (1992).
146. Jahn, R., Schiebler, W., Ouimet, C. & Greengard, P. A 38,000-dalton membrane protein (p38) present in synaptic vesicles. Proc Natl Acad Sci U S A 82, 4137-4141 (1985).
147. Wiedenmann, B. & Franke, W. W. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell 41, 1017-1028 (1985).
148. Leube, R. E. et al. Synaptophysin: molecular organization and mRNA expression as determined from cloned cDNA. Embo j 6, 3261-3268 (1987).
149. Südhof, T. C., Lottspeich, F., Greengard, P., Mehl, E. & Jahn, R. A synaptic vesicle protein with a novel cytoplasmic domain and four transmembrane regions. Science 238, 1142-1144 (1987).
150. Jahn, R. & Südhof, T. C. Synaptic vesicles and exocytosis. Annu Rev Neurosci 17, 219-246 (1994).
151. Martineau, M. et al. Storage and uptake of D-serine into astrocytic synaptic-like vesicles specify gliotransmission. J Neurosci 33, 3413-3423 (2013).
152. Bradberry, M. M. et al. Rapid and Gentle Immunopurification of Brain Synaptic Vesicles. J Neurosci 42, 3512-3522 (2022).
153. Ahmed, S., Holt, M., Riedel, D. & Jahn, R. Small-scale isolation of synaptic vesicles from mammalian brain. Nat Protoc 8, 998-1009 (2013).
154. Takamori, S. et al. Molecular anatomy of a trafficking organelle. Cell 127, 831-846 (2006).
155. Geppert, M. et al. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717-727 (1994).
156. Link, E. et al. Tetanus toxin action: inhibition of neurotransmitter release linked to synaptobrevin proteolysis. Biochem Biophys Res Commun 189, 1017-1023 (1992).
157. Schiavo, G. et al. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359, 832-835 (1992).
158. Bajjalieh, S. M., Peterson, K., Shinghal, R. & Scheller, R. H. SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science 257, 1271-1273 (1992).
159. Cidon, S. & Sihra, T. S. Characterization of a H+-ATPase in rat brain synaptic vesicles. Coupling to L-glutamate transport. J Biol Chem 264, 8281-8288 (1989).
160. Takamori, S., Rhee, J. S., Rosenmund, C. & Jahn, R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407, 189-194 (2000).
161. McIntire, S. L., Reimer, R. J., Schuske, K., Edwards, R. H. & Jorgensen, E. M. Identification and characterization of the vesicular GABA transporter. Nature 389, 870-876 (1997).
162. Kunii, M. et al. SNAP23 deficiency causes severe brain dysplasia through the loss of radial glial cell polarity. J Cell Biol 220, e201910080 (2021).
163 . Ravichandran, V., Chawla, A. & Roche, P. A. Identification of a novel syntaxin- and synaptobrevin/VAMP-binding protein, SNAP-23, expressed in non-neuronal tissues. J Biol Chem 271, 13300-13303 (1996).
164. Suh, Y. H. et al. A neuronal role for SNAP-23 in postsynaptic glutamate receptor trafficking. Nature Neuroscience13, 338-U316 (2010).
165. Woods, D. F. & Bryant, P. J. The discs-large tumor suppressor gene of Drosophila encodes a guanylate kinase homolog localized at septate junctions. Cell 66, 451-464 (1991).
166. Cho, K. O., Hunt, C. A. & Kennedy, M. B. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron 9, 929-942 (1992).
167. Nakamura, N. et al. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol 131, 1715-1726 (1995).
168. Griffith, K. J. et al. Molecular cloning of a novel 97-kd Golgi complex autoantigen associated with Sjögren's syndrome. Arthritis Rheum 40, 1693-1702 (1997).
169. Mu, F. T. et al. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine "fingers" and contains a calmodulin-binding IQ motif. J Biol Chem 270, 13503-13511 (1995).
170. Tie, C., Zhang, D. W., Chen, H. X., Song, S. L. & Zhang, X. X. Study of the electrical connection mechanism of sheathless interface for capillary electrophoresis-electrospray ionization-mass spectrometry. J Mass Spectrom 47, 1429-1434 (2012).
171. Meyer, H. S. et al. Inhibitory interneurons in a cortical column form hot zones of inhibition in layers 2 and 5A. Proc Natl Acad Sci U S A 108, 16807-16812 (2011).
172. Olbrich, H. G. & Braak, H. Ratio of pyramidal cells versus non-pyramidal cells in sector CA1 of the human Ammon's horn. Anat Embryol (Berl) 173, 105-110 (1985).
173. Tremblay, R., Lee, S. & Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 91, 260-292 (2016).
174. Li, X. et al. Generation of a whole-brain atlas for the cholinergic system and mesoscopic projectome analysis of basal forebrain cholinergic neurons. Proc Natl Acad Sci U S A 115, 415-420 (2018).
175. Herculano-Houzel, S., Mota, B. & Lent, R. Cellular scaling rules for rodent brains. Proc Natl Acad Sci U S A 103, 12138-12143 (2006).
176. Ishimura, K. et al. Quantitative analysis of the distribution of serotonin-immunoreactive cell bodies in the mouse brain. Neurosci Lett 91, 265-270 (1988).
177. Egashira, Y. et al. Unique pH dynamics in GABAergic synaptic vesicles illuminates the mechanism and kinetics of GABA loading. Proc Natl Acad Sci U S A 113, 10702-10707 (2016).
178. Mani, M. & Ryan, T. A. Live imaging of synaptic vesicle release and retrieval in dopaminergic neurons. Front Neural Circuits 3, 3 (2009).
179. Egashira, Y., Takase, M. & Takamori, S. Monitoring of vacuolar-type H+ ATPase-mediated proton influx into synaptic vesicles. J Neurosci 35, 3701-3710 (2015).
180. Qian, C. et al. Localization, proteomics, and metabolite profiling reveal a putative vesicular transporter for UDP-glucose. Elife 10, e65417 (2021).
181. Schenck, S., Wojcik, S. M., Brose, N. & Takamori, S. A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat Neurosci 12, 156-162 (2009).
182. Duran-Trio, L. et al. A new rat model of creatine transporter deficiency reveals behavioral disorder and altered brain metabolism. Sci Rep 11, 1636 (2021).
183. Stockebrand, M. et al. A Mouse Model of Creatine Transporter Deficiency Reveals Impaired Motor Function and Muscle Energy Metabolism. Front Physiol 9, 773 (2018).
184. Guthmiller, P., Van Pilsum, J. F., Boen, J. R. & McGuire, D. M. Cloning and sequencing of rat kidney L-arginine:glycine amidinotransferase. Studies on the mechanism of regulation by growth hormone and creatine. J Biol Chem 269, 17556-17560 (1994).
185. Skarnes, W. C. et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337-342 (2011).
186. Kolodziej, P. A. & Young, R. A. Epitope tagging and protein surveillance. Methods Enzymol 194, 508-519 (1991).
187. Ryan, M. D., King, A. M. & Thomas, G. P. Cleavage of foot-and-mouth disease virus polyprotein is mediated by residues located within a 19 amino acid sequence. J Gen Virol 72 ( Pt 11), 2727-2732 (1991).
188. Ahier, A. & Jarriault, S. Simultaneous expression of multiple proteins under a single promoter in Caenorhabditis elegans via a versatile 2A-based toolkit. Genetics 196, 605-613 (2014).
189. Daniels, R. W., Rossano, A. J., Macleod, G. T. & Ganetzky, B. Expression of multiple transgenes from a single construct using viral 2A peptides in Drosophila. PLoS One 9, e100637 (2014).
190. Sauer, B. & Henderson, N. Cre-stimulated recombination at loxP-containing DNA sequences placed into the mammalian genome. Nucl Acids Res 17, 147-161 (1989).
191. Gu, H., Zou, Y. R. & Rajewsky, K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell 73, 1155-1164 (1993).
192. Feil, R., Wagner, J., Metzger, D. & Chambon, P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun 237, 752-757 (1997).
193. Indra, A. K. et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucl Acids Res 27, 4324-4327 (1999).
194. Hamberger, A., Chiang, G. H., Sandoval, E. & Cotman, C. W. Glutamate as a CNS transmitter. II. Regulation of synthesis in the releasable pool. Brain Res 168, 531-541 (1979).
195. Hamberger, A. C., Chiang, G. H., Nylen, E. S., Scheff, S. W. & Cotman, C. W. Glutamate as a CNS transmitter. I. Evaluation of glucose and glutamine as precursors for the synthesis of preferentially released glutamate. Brain Res 168, 513-530 (1979).
196. McBride, W. J., Flint, R. S., Ciancone, M. T. & Murphy, J. M. In vitro release of endogenous monoamines and amino acids from several CNS regions of the rat. Neurochem Res 8, 245-257 (1983).
197. Nadler, J. V., White, W. F., Vaca, K. W., Redburn, D. A. & Cotman, C. W. Characterization of putative amino acid transmitter release from slices of rat dentate gyrus. J Neurochem 29, 279-290 (1977).
198. Keith, R. A., Horn, M. B., Piser, T. M. & Mangano, T. J. Effects of stimulus intensity on the inhibition by omega-conotoxin GVIA and neomycin of K(+_-evoked [3H]norepinephrine release from hippocampal brain slices and synaptosomal calcium influx. Biochem Pharmacol 45, 165-171 (1993).
199. Eliseeva, Z. V. & Durinian, R. A. [Projections of the somatosensory areas of the cerebral cortex into the thalamic nuclei]. Biull Eksp Biol Med 80, 113-117 (1975).
200 . Yamawaki, N., Raineri Tapies, M. G., Stults, A., Smith, G. A. & Shepherd, G. M. Circuit organization of the excitatory sensorimotor loop through hand/forelimb S1 and M1. Elife 10, e66836 (2021).
201. Scala, F. et al. Layer 4 of mouse neocortex differs in cell types and circuit organization between sensory areas. Nat Commun 10, 4174 (2019).
202. Stumpf, A. et al. Cannabinoid type 2 receptors mediate a cell type-specific self-inhibition in cortical neurons. Neuropharmacology 139, 217-225 (2018).
203. Gulyassy, P. et al. Proteomic comparison of different synaptosome preparation procedures. Amino Acids 52, 1529-1543 (2020).
204. Antonucci, F. et al. SNAP-25, a Known Presynaptic Protein with Emerging Postsynaptic Functions. Front Synaptic Neurosci 8, 7 (2016).
205. Cserép, C. et al. NMDA receptors in GABAergic synapses during postnatal development. PLoS One 7, e37753 (2012).
206. Cameron, R., Klein, L., Shyjan, A. W., Rakic, P. & Levenson, R. Neurons and astroglia express distinct subsets of Na,K-ATPase alpha and beta subunits. Brain Res Mol Brain Res 21, 333-343 (1994).
207. Mersel, M., Lelong, I., Hindelang, C., Sarlieve, L. & Vincendon, G. Isolation of plasma membranes from neurons grown in primary culture. Anal Biochem 166, 246-252 (1987).
208. Sun, G. Y., Huang, H. M., Kelleher, J. A., Stubbs, E. B., Jr. & Sun, A. Y. Marker enzymes, phospholipids and acyl group composition of a somal plasma membrane fraction isolated from rat cerebral cortex: a comparison with microsomes and synaptic plasma membranes. Neurochem Int 12, 69-77 (1988).
209. Schrimpf, S. P. et al. Proteomic analysis of synaptosomes using isotope-coded affinity tags and mass spectrometry. Proteomics 5, 2531-2541 (2005).
210. Fykse, E. M. & Fonnum, F. Uptake of Gamma-Aminobutyric Acid by a Synaptic Vesicle Fraction Isolated from Rat-Brain. J Neurochem 50, 1237-1242 (1988).
211. Bellocchio, E. E., Reimer, R. J., Fremeau, R. T., Jr. & Edwards, R. H. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289, 957-960 (2000).
212. Oja, S. S. & Saransaari, P. Taurine and neurotransmission. In Taurine in Health and Disease, Ed (El Idrissi, A., and L’Amoreaux, W.). (Trivandrum, Kerala, 2012).
213. D'Aniello, S., Somorjai, I., Garcia-Fernàndez, J., Topo, E. & D'Aniello, A. D-Aspartic acid is a novel endogenous neurotransmitter. Faseb J 25, 1014-1027 (2011).
214. Abe, K. & Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16, 1066-1071 (1996).
215. Reis, D. J. & Regunathan, S. Is agmatine a novel neurotransmitter in brain? Trends Pharmacol Sci 21, 187-193 (2000).
216. Misu, Y., Goshima, Y. & Miyamae, T. Is DOPA a neurotransmitter? Trends Pharmacol Sci 23, 262-268 (2002).
217. Balthazart, J. & Ball, G. F. Is brain estradiol a hormone or a neurotransmitter? Trends Neurosci 29, 241-249 (2006).
218. Tiedje, K. E., Stevens, K., Barnes, S. & Weaver, D. F. Beta-alanine as a small molecule neurotransmitter. Neurochem Int 57, 177-188 (2010).
219. Du, J. et al. Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proc Natl Acad Sci U S A 111, 8961-8966 (2014).
220. Almeida, L. S., Salomons, G. S., Hogenboom, F., Jakobs, C. & Schoffelmeer, A. N. Exocytotic release of creatine in rat brain. Synapse 60, 118-123 (2006).
221. Peral, M. J., Vazquez-Carretero, M. D. & Ilundain, A. A. Na+/Cl-/CREATINE TRANSPORTER ACTIVITY AND EXPRESSION IN RAT BRAIN SYNAPTOSOMES. Neuroscience 165, 53-60 (2010).
222. Cooper, J. R., Bloom, F. E. & Roth, R. H. The Biochemical Basis of Neuropharmacology p.2. (Oxford University Press, 2003).
223. Squire, L., Berg, D., Bloom, F.E., du Lac, S., Ghosh, A., and Spitzer, N.C. Fundamental Neuroscience, 4thedition. (Elsevier, 2012).
224. Robinson, J. D. Mechanisms of Synaptic Transmission. Bridging the Gaps. (Oxford University Press, 2001).
225. Barañano, D. E., Ferris, C. D. & Snyder, S. H. Atypical neural messengers. Trends Neurosci 24, 99-106 (2001).
226. Brown, D. A. & Scholfield, C. N. Depolarization of neurones in the isolated olfactory cortex of the guinea-pig by gamma-aminobutyric acid. Br J Pharmacol 65, 339-345 (1979).
227. Pringle, A. K., Gardner, C. R. & Walker, R. J. Reduction of cerebellar GABAA responses by interleukin-1 (IL-1) through an indomethacin insensitive mechanism. Neuropharmacology 35, 147-152 (1996).
228. Osmanović, S. S. & Shefner, S. A. gamma-Aminobutyric acid responses in rat locus coeruleus neurones in vitro: a current-clamp and voltage-clamp study. J Physiol 421, 151-170 (1990).
229. Akaike, A., Ohno, Y., Sasa, M. & Takaori, S. Excitatory and inhibitory effects of dopamine on neuronal activity of the caudate nucleus neurons in vitro. Brain Res 418, 262-272 (1987).
230. Cole, A. E., ffench-Mullen, J. M. & Fisher, R. S. Fade of the response to prolonged glutamate application in the rat hippocampal slice. Synapse 4, 11-18 (1989).
231 . Beck, S. G. & Goldfarb, J. Serotonin produces a reversible concentration dependent decrease of population spikes in rat hippocampal slices. Life Sci 36, 557-563 (1985).
232. Reynolds, J. N., Baskys, A. & Carlen, P. L. The effects of serotonin on N-methyl-D-aspartate and synaptically evoked depolarizations in rat neocortical neurons. Brain Res 456, 286-292 (1988).
233. Mendiguren, A., Aostri, E., Alberdi, E., Pérez-Samartín, A. & Pineda, J. Functional characterization of cannabidiol effect on the serotonergic neurons of the dorsal raphe nucleus in rat brain slices. Front Pharmacol 13, 956886 (2022).
234. Xie, D. J. et al. Identification of 5-HT receptor subtypes enhancing inhibitory transmission in the rat spinal dorsal horn in vitro. Mol Pain 8, 58 (2012).
235. Gorelova, N. & Reiner, P. B. Role of the afterhyperpolarization in control of discharge properties of septal cholinergic neurons in vitro. J Neurophysiol 75, 695-706 (1996).
236. Okuhara, D. Y. & Beck, S. G. 5-HT1A receptor linked to inward-rectifying potassium current in hippocampal CA3 pyramidal cells. J Neurophysiol 71, 2161-2167 (1994).
237. Sizer, A. R., Kilpatrick, G. J. & Roberts, M. H. A post-synaptic depressant modulatory action of 5-hydroxytryptamine on excitatory amino acid responses in rat entorhinal cortex in vitro. Neuropharmacology 31, 531-539 (1992).
238. De Deyn, P. P. & Macdonald, R. L. Guanidino compounds that are increased in cerebrospinal fluid and brain of uremic patients inhibit GABA and glycine responses on mouse neurons in cell culture. Ann Neurol 28, 627-633 (1990).
239. De Deyn, P. P., D'Hooge, R., Van Bogaert, P. P. & Marescau, B. Endogenous guanidino compounds as uremic neurotoxins. Kidney Int Suppl 78, S77-83 (2001).
240. Neu, A. et al. Activation of GABA(A) receptors by guanidinoacetate: a novel pathophysiological mechanism. Neurobiol Dis 11, 298-307 (2002).
241. Koga, Y. et al. Brain creatine functions to attenuate acute stress responses through GABAnergic system in chicks. Neuroscience 132, 65-71 (2005).
242. Royes, L. F. et al. Neuromodulatory effect of creatine on extracellular action potentials in rat hippocampus: role of NMDA receptors. Neurochem Int 53, 33-37 (2008).
243. Braissant, O. & Henry, H. AGAT, GAMT and SLC6A8 distribution in the central nervous system, in relation to creatine deficiency syndromes: a review. J Inherit Metab Dis 31, 230-239 (2008).
244. Braissant, O., Béard, E., Torrent, C. & Henry, H. Dissociation of AGAT, GAMT and SLC6A8 in CNS: relevance to creatine deficiency syndromes. Neurobiol Dis 37, 423-433 (2010).
245. Dunbar, M., Jaggumantri, S., Sargent, M., Stockler-Ipsiroglu, S. & van Karnebeek, C. D. Treatment of X-linked creatine transporter (SLC6A8) deficiency: systematic review of the literature and three new cases. Mol Genet Metab 112, 259-274 (2014).
246. Mercimek-Mahmutoglu, S. et al. Treatment of intractable epilepsy in a female with SLC6A8 deficiency. Mol Genet Metab 101, 409-412 (2010).
247. Gerbatin, R. R. et al. Delayed creatine supplementation counteracts reduction of GABAergic function and protects against seizures susceptibility after traumatic brain injury in rats. Prog Neuro-Psychopharmacol Biol Psych 92, 328-338 (2019).
248. Braissant, O. Creatine and guanidinoacetate transport at blood-brain and blood-cerebrospinal fluid barriers. J Inherit Metab Dis 35, 655-664 (2012).
249 Brosnan, M. E. & Brosnan, J. T. The role of dietary creatine. Amino Acids 48, 1785-1791 (2016).
250. Watanabe, A., Kato, N. & Kato, T. Effects of creatine on mental fatigue and cerebral hemoglobin oxygenation. Neurosci Res 42, 279-285 (2002).
251 . Rae, C., Digney, A. L., McEwan, S. R. & Bates, T. C. Oral creatine monohydrate supplementation improves brain performance: a double-blind, placebo-controlled, cross-over trial. Proc Biol Sci 270, 2147-2150 (2003).
252. McMorris, T. et al. Effect of creatine supplementation and sleep deprivation, with mild exercise, on cognitive and psychomotor performance, mood state, and plasma concentrations of catecholamines and cortisol. Psychopharmacology (Berl) 185, 93-103 (2006).
253. McMorris, T. et al. Creatine supplementation, sleep deprivation, cortisol, melatonin and behavior. Physiol Behav 90, 21-28 (2007).
254. Rae, C. D. & Bröer, S. Creatine as a booster for human brain function. How might it work? Neurochem Int 89, 249-259, (2015).
255. Wallimann, T. & Harris, R. Creatine: a miserable life without it. Amino Acids 48, 1739-1750 (2016).
256. Andreassen, O. A. et al. Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Dis 8, 479-491 (2001).
257. Andres, R. H. et al. Effects of creatine treatment on the survival of dopaminergic neurons in cultured fetal ventral mesencephalic tissue. Neuroscience 133, 701-713 (2005).
258. Paxinos, G. & Franklin., K. B. J. Paxinos and Franklin's the mouse brain in stereotaxic coordinates 4th edition. (Elsevier/Academic Press, 2013).
259. Booth, R. F. G. & Clark, J. B. Rapid Method for Preparation of Relatively Pure Metabolically Competent Synaptosomes from Rat-Brain. Biochem J 176, 365 (1978).
260. Hell, J. W., Maycox, P. R., Stadler, H. & Jahn, R. Uptake of GABA by rat brain synaptic vesicles isolated by a new procedure. Embo J 7, 3023-3029 (1988).
261. Schmidt, A. et al. Severely altered guanidino compound levels, disturbed body weight homeostasis and impaired fertility in a mouse model of guanidinoacetate N-methyltransferase (GAMT) deficiency. Hum Mol Genet 13, 905-921 (2004).
262. Rosko, L. M. et al. Cerebral Creatine Deficiency Affects the Timing of Oligodendrocyte Myelination. J Neurosci 43, 1143-1153 (2023).
263. Philibert, R. A., Rogers, K. L. & Dutton, G. R. K+-evoked taurine efflux from cerebellar astrocytes: on the roles of Ca2+ and Na+. Neurochem Res 14, 43-48 (1989).
264. Saransaari, P. & Oja, S. S. Characteristics of taurine release in slices from adult and developing mouse brain stem. Amino Acids 31, 35-43 (2006).
265. Takuma, K. et al. Ca2+ depletion facilitates taurine release in cultured rat astrocytes. Jpn J Pharmacol 72, 75-78 (1996).
266. Molchanova, S. M., Oja, S. S. & Saransaari, P. Mechanisms of enhanced taurine release under Ca2+ depletion. Neurochem Int 47, 343-349 (2005).
267. Ghirardini, E., G. Sagona, A. Marquez-Galera, F. Calugi, C. M. Navarron, F. Cacciante, S. Chen, F. Di Vetta, L. Dada, R. Mazziotti, L. Lupori, E. Putignano, P. Baldi, J. P. Lopez-Atalaya, T. Pizzorusso, and L. Baroncelli. . Cell-specific vulnerability to metabolic failure: the crucial role of parvalbumin expressing neurons in creatine transporter deficiency. Acta Neuropathol Commun 11,33 (2023).